Day 1 :
Keynote Forum
Boris Y Zaslavsky
Cleveland Diagnostics, USA
Keynote: Properties of membrane-less organelles and aqueous two-phase systems
Time : 9:35-10:05

Biography:
Boris Y Zaslavsky is a Bioanalytical Chemist. He graduated from Moscow State University in 1967, PhD in 1972; DSc in 1985 from USSR Academy of Sciences, Moscow, USSR. From 2012-present, he is a Chief Scientific Officer of Cleveland Diagnostics, Cleveland, OH, from 1997-present, he is a Vice President, Director of Research and Cofounder of Analiza, Inc., Cleveland, OH. He has over 180 publications in peer-reviewed journals, 1 monograph, over 10 USA and international patents issued. His research interests are: Clinical proteomics, role of water in biology, and aqueous two-phase systems.
Abstract:
Coordination of numerous cellular biochemical reactions in space and time is achieved by compartmentalization. In addition to intracellular membranes acting as physical barriers for several cellular organelles there is a multitude of membrane-less organelles formed by liquid-liquid phase separation. The principles governing phase separation and functions of such organelles in vivo are poorly understood as of now. However, the much better studied aqueous two-phase systems formed by two polymers may serve as a model of membrane-less organelles. Such systems originate from polymer influence on the solvent properties of water. The phase forming polymers may include proteins and polysaccharides. The differences between solvent features of aqueous media in the two phases may be quantified and manipulated by polymers’ concentrations and additives of inorganic salts or small organic compounds, such as sucrose, sorbitol, etc. The differences between electrostatic properties of the phases as well as those between solvent features may be quantified using partitioning of homologous series of charged compounds and solvatochromic dyes as molecular probes for the solvent dipolarity/polarizability, solvent H-bond donor acidity, and solvent H-bond acceptor basicity. The differences between solvent features and electrostatic properties of the phases govern unequal distribution of proteins and other natural compounds in aqueous two phase systems and in membrane-less organelles. This solvent-driven partitioning, and not the “normal” protein-protein interactions, might cause enrichment of some proteins within the membrane-less organelles. It will be shown that proteins may influence solvent features of water and their effects are similar or exceeding those displayed by common macromolecular crowding agents and organic osmolytes. It is suggested that the effects of proteins on the solvent features of aqueous media may regulate the phase separation in vivo.
Keynote Forum
Bin Huang
Kaohsiung Medical University, Taiwan
Keynote: Integrative analysis of post-translational protein S-nitrosylation in endothelial cells
Time : 10:05-10:35

Biography:
Bin Huang obtained his PhD degree from the Department of Plant Science, National Taiwan University. He then focused on Cardiology during his Postdoctoral studies. He became experienced in the gaseous molecule-mediated post-translational proteome, particularly in the NO-mediated S-nitrosylation when studying endothelial cells of the vascular system. He became interested in the behavior of mitochondrial fusion/fission when studying cell aging and drug-resistance of cancer cells. In addition to these research interests, he develops the Center for Stem Cell Research of Kaohsiung Medical University as the Vice Director.
Abstract:
Nitric oxide (NO), an endogenous evolutionary gaseous molecule with labile character can bind to cysteine residues (Cys-NO, S-nitrosylation) and then alter the enzyme activity. NO is regarded as a mild reactive oxygen/nitrogen species (ROS/RNS) that can compete with other, more potent ROS/RNS, and protects cells from irreversible oxidative stress caused by free radicals. At present available methodologies applied to study the implications of NO in physiological responses include Western blotting to measure the phosphorylation of endothelial nitric oxide synthase (eNOS) at Ser1177 and Ser633 residues and detecting gaseous NO by Griess reagent. However, this reagent is greatly affected by the presence of peroxinitrite (ONOO−). Therefore, the new fluorescent probe - 5-amino-2-(6-hydroxy-3-oxo-3H-xanthen-9-yl) benzoic acid methylester (FA-OMe) - that specifically binds to endogenous NO, was developed and utilized. As a result, the elevated production of NO can be estimated not only by eNOS phosphorylation in Western blots but also by direct quantification using FA-OMe. Once it became possible to confirm the production of NO, the identification of the subsequent protein S-nitrosylation resulted from NO binding to the cysteine residues became important. The utilizations of commercialized antibody and mass spectrometric devices were reported to detect the Cys-NO residue directly. However, for the reason of poor antibody specificity and weak chemical binding of Cys-NO, both two methods were not reliable. We therefore designed a tag-based labeling system on cysteine residue that modified from biotin-switch, (e.g. IAA, IAM and iTRAQ). Cys-NO will be replaced by these tags and was then detected either by 2-DE-based Western blot or mass spectrometry with identical molecular weight shifts. The whole profiles of enzyme activation, gaseous NO molecule production, and the subsequent protein S-nitrosylation could be analyzed simultaneously to explain more details about the physiological mechanisms of action in protein S-nitrosylation.
Keynote Forum
Manuel Gea
Bio-Modeling Systems, France
Keynote: The future of systems medicine: Everything you always wanted to know about the “reality†of big data and AI “big mirages†but were afraid to ask
Time : 10:55-11:25

Biography:
Manuel Gea is a Serial Entrepreneur, Co-Founder of the world’s first Mechanisms-Based Medicine Company, thinking & doing out of the box applying “general semantics” principles and Augmented Intelligence vs. Artificial Intelligence. He is a Keynote Speaker, independent board member, expert & business angel developing disruptive innovations (technologies, novel therapies & business models) in the life sciences, IT, pharma, healthcare & cosmetic sectors. He is graduated from Ecole Centrale Paris in operational research and data integration, and has Sociology of Organizations degree from Paris IX Dauphine University. He is also Chairman of the Independent trans-discipline biotech Think Tank Adebiotech, and the Co-founder and Chairman of Centrale-Santé, the French Health Think-Tank gathering 2500 members, professional involved in sector innovations and creating value for patients. He is Co-founder, CEO & VP R&D IT of BMSystems, dedicated to the discovery of cost-effective new therapeutic, diagnostic & preventive solutions.
Abstract:
Statement of the Problem: With a 95% failure rate, the Big Pharma R&D model focused on testing new patentable compounds for novel targets based on KOL concepts is not more pertinent for at least 3 reasons: 1) Despite decades of investments in Omics technologies and Systems Biology programs produced few relevant results due to 3 “side effects” and a conceptual mistake: Life mechanisms are complex not complicated! 2) The “mirage” of Artificial Intelligence (AI) that MUST follow rules in a world where humans massively do not. 3) The unreliability of scientific and clinical publications is increasing, and the valuable negative results are not published. Why the transfer of the digital tools and methods does from the internet world to the life sciences R&D world does not work properly. One reason is that the founding basements of the two worlds do not obey the same rules. The challenge is not a question of technologies only, its needs to redefine discovery concepts.
The Future of Systems Medicine: 1) Understanding and validating the mechanisms of a disease/disorder becomes the first objective. 2) Finding the most adapted solutions is a necessary consequence. The Mechanisms-Based Medicine Concept gives the basements to apply CADI™ (Computer Augmented Deductive Intelligence) Discovery that addresses life mechanisms complexity and the unreliability of scientific and clinical publications by combining the strengths of human and artificial intelligences in the right order.
The 5 CADI™ Discovery Principles: 1) Mechanisms-Based Medicine Concept; 2) Architectural Principle; 3) Negative Selection Principle; 4) Steps Validation Principle; 5) Integrated Solutions Principles.
Conclusions: CADI Discovery already led to a world’s first in neurodegenerative diseases, 1 therapeutic spin-off and 1 exclusive our license, 4 issued patents, 10 publications, and 39 CADI™ programs among completed or open for collaboration.

Recent Publications:
1. F Iris F, D Filiou, Ch W Turck (2014) Differential proteomics analyses reveal anxiety-associated molecular and cellular mechanisms in cingulate cortex synapses. American Journal of Psychiatry and Neuroscience 2(3): 25-42.
2. Iris F (2012) Psychiatric systems medicine: closer at hand than anticipated but not with the expected portrait. Pharmacopsychiatry 45 (Suppl. 1): S12–S2.
3. Turck CW, Iris F (2011) Proteome-based pathway modelling of psychiatric disorders. Pharmacopsychiatry 44 (Suppl 1): S54-61.
4. Iris F, Gea M, Lampe PH and Santamaria P (2009) Production and implementation of predictive biological models. Med Sci. 25: 608-16.
5. Iris F, Gea M, Lampe PH and Santamaria P (2009) Production and implementation of predictive biological models. Med Sci. 25: 608-16.
- Mass Spectrometry in Proteome Research | Protein Expression and Analysis | Proteomics from Discovery to Function | Molecular Medicine and Diagnostics
Location: Paris, France
Session Introduction
Oliva Petra
Sanofi Genzyme, USA
Title: Strategy for determining clinical biomarker panel
Time : 11:25-11:45

Biography:
Petra Oliva has 10 years of biotech experience and close to 20 years scientific expertise in an advanced interdisciplinary research in analytical chemistry, biotechnology and biochemistry with deep understanding of biomarker discovery, qualification and validation. She is currently Pr. Scientist at Translational Science group within Sanofi, US. She is managing analytical/bioanalytical support from pre-clinical stage, through PhI & PhII all the way to the commercial phase. Petra and her team established several innovation platforms (as targeted proteomics & MS imaging) winning multiple global Sanofi awards. She also has 8 years collaboration with CDC on LSD newborn screening.
Abstract:
Biomarkers are essential for improving the outcomes of clinical trials and accelerating drug development. Mass spectrometry (MS)-based proteomics applied early to clinical samples has the potential to identify and narrow down predictive and pharmacodynamics biomarkers. These markers can then be used in clinical trials for patient stratification and to increase sensitivity of primary endpoints for a better measurement of therapeutic response. Unbiased proteomic profiling is powerful during the exploratory biomarker stage for monitoring hundreds or thousands of proteins, but throughput is low and relative quantitation is variable for low abundant analytes. Here, we describe an integrated, hypothesis-driven strategy that combines unbiased proteomics and literature mining to generate a highly quantitative and reproducible targeted proteomics assay for testing in large, representative patient cohorts for candidate biomarker screening. Combined with appropriate statistical and bioinformatics processes, this strategy will facilitate selection of a robust biomarker panel which may be validated as a companion diagnostic or as a clinical tool.
Ida Chiara Guerrera
INSERM, France
Title: Proteomics of rare genetic diseases: Impact of cystinosin mutations on protein stability and protein network
Time : 11:45-12:05

Biography:
Ida Chiara Guerrera (PhD, UCL London) is Head of the Proteomics Laboratory at the Necker Hospital in Paris. Her interests center on understanding human disease by applying innovative proteomics approaches to biomedical research. She participates in fundamental research to understand mechanisms underlying disease and translational research for biomarker discovery. Her work has contributed to exploring three rare genetic diseases: cystic fibrosis, cystinurie and cystinosis.
Abstract:
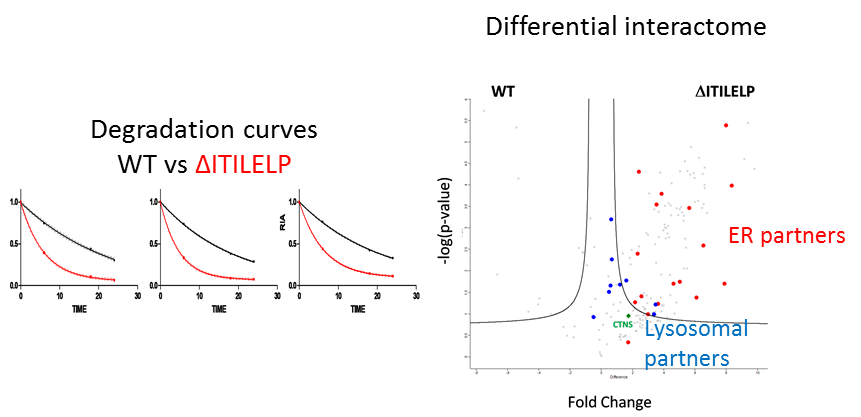
Statement of the Problem: Cystinosis is a rare autosomal recessive storage disorder characterized by defective lysosomal efflux of cystine due to mutations in the CTNS gene encoding the lysosomal cystine transporter, cystinosin. Over 100 mutations have been reported, leading to varying disease severity, often in correlation with residual cystinosin activity as a transporter. However, the correlation between genotype and phenotype is not always clear and we applied different proteomics approaches to better understand the mechanisms of this disease. To unveil additional roles of cystinosin, we studied the protein interaction of the WT cystinosin and different mutations. Furthermore, we focused on an atypical mutation concerning protein glycolsilation, ΔITILELP, that sometimes leads to severe forms.
Methodology & Theoretical Orientation: Co-immunoprecipitation and dynamic SILAC were used. All analyses were performed on a nanoRSLC Q-Exactive Plus MS.
Findings: We found cystinosin interacts with almost all components of vacuolar H(+)-ATPase and the Ragulator complex and with the small GTPases Ras-related GTP-binding protein A (RagA) and RagC. These interactions are lost when cystinosin carries severe loss-of-activity mutations. We also showed that wild-type cystinosin is very stable, while ΔITILELP is degraded three times more rapidly. We observed that in the lysosome, ΔITILELP is still capable of interacting with the V-ATPase complex and some members of the mTOR pathway, similar to the wild-type protein. Our interactomic and immunofluorescence studies showed that ΔITILELP is partially retained at the endoplasmic reticulum (ER).
Conclusion & Significance: Our results show a dual role for cystinosin as a cystine transporter and as a component of the mTORC1 pathway, and provide an explanation for the appearance of Fanconi syndrome in cystinosis. We also found that the high turnover of ΔITILELP, due to its immature glycosylation state in combination with low transport activity, might be responsible for the phenotype observed in some patients.
Recent Publications
1. Nevo N (2017) Impact of cystinosin mutations on protein stability by differential dynamic SILAC. Molecular and Cellular Proteomics (In press).
2. Lipecka J (2016) Sensitivity of mass spectrometry analysis depends on the shape of the filtration unit used for filter aided sample preparation (FASP). Proteomics. 16(13):1852-7.
3. Chhuon C (2016) Changes in lipid raft proteome upon TNF-α stimulation of cystic fibrosis cells. J. Proteomics. 145:246-53.
4. Andrzejewska Z (2016) Cystinosin is a component of the vacuolar H+-ATPase-Ragulator-Rag complex controlling mammalian target of rapamycin complex 1 signaling. J Am Soc Nephrol. 27(6):1678-88.
5. Ramond E (2015) Importance of host cell arginine uptake in Francisella phagosomal escape and ribosomal protein amounts. Mol Cell Proteomics. 14(4):870-81.
Marcus Krüger
University of Cologne, Germany
Title: Dynamic changes in the skeletal muscle proteome during denervation-induced atrophy
Time : 12:05-12:25

Biography:
Marcus Krüger studies Chemistry and received his PhD in Natural Sciences from the Univeristy of Halle-Wittenberg, working on basic-helix-loop-helix transcription factors in mouse. He did his Postdoctoral work at the Syddansk University in Odense working on quantitative mass spectrometry and stable isotope labeling of living animals. Since moving to the MPI for Heart and Lung Research in 2008, he became a Service Group Leader and in 2014 he was recruited as a Principle Investigator to the Institute for Genetics in Cologne. His current research interests includes quantitative mass spectrometryand the enrichment of posttranslational modifications in both health and disease related conditons.
Abstract:
Loss of neuronal stimulation enhances protein breakdown and reduces protein synthesis, causing rapid muscle mass loss. To elucidate the pathophysiological adaptations that occur in atrophying muscles, we used stable isotope labelling and mass spectrometry to accurately quantify protein expression changes during denervation-induced atrophy after sciatic nerve section in skeletal muscle. Additionally, mice were fed a SILAC diet containing 13C6 lysine for four, seven, or eleven days to calculate relative levels of protein synthesis in denervated and control muscles. Ubiquitin remnant peptides (K-e-GG) were profiled by immunoaffinity enrichment to identify potential substrates of the ubiquitin proteasomal pathway. Besides a protein expression profiling we used a pulse-SILAC labelling approach to identify differential Lys6 incorporation rates between control and denervated muscle. Enrichment of diglycine remnants identified 2100 endogenous ubiquitination sites and revealed a metabolic and myofibrillar protein diglycine signature, including myosin heavy chains (MyHC), myomesins and titin, during denervation. Comparative analysis of these proteomic datasets with known atrogenes using a random forest approach identified 92 proteins subject to atrogene-like regulation that have not previously been directly associated with denervation-induced atrophy. Comparison of protein synthesis and proteomic data indicated upregulation of specific proteins in response to denervation is mainly achieved by protein stabilization. This study provides the first integrated analysis of protein expression, synthesis and ubiquitin signatures during muscular atrophy in a living animal.
Jean-Michel Camadro
Institut Jacques Monod, France
Title: High-sensitivity, high-resolution MS-based quantitative proteomics by manipulation of protein isotope
Time : 12:25-12:45

Biography:
Abstract:
Many quantitative proteomics strategies rely on in vivo metabolic incorporation into proteins of amino acids with modified stable isotopes profiles. These methods give rise to multiple ions for each peptide, with possible distortion of the isotopologs distribution, making the overall analytical process complex. By reducing the isotopic composition of proteins in vivo, we bring a new dimension to in-depth, high resolution MS-based quantitative proteomics that alleviates these problems. We used U-[12C]-glucose as the metabolic precursor of all amino acids in yeast. This substantially increased the peptide monoisotopic ion intensity in bottom-up analyses, greatly improving identification scores and protein sequence coverage. Multiplexing samples of 12C composition varying from natural abundance to 100% 12C makes it possible to address quantitative proteomics, keeping all the critical information within a single isotopologs cluster. We applied this method to measure for the first time protein turnover at the proteome scale in the pathogenic yeast Candida albicans.
Bin Huang
Kaohsiung Medical University, Taiwan
Title: Application of embryogenic mouse as a platform in identifying human missing proteins
Time : 12:45-13:05

Biography:
Bin Huang gained his PhD degree from Department of Plant Science, National Taiwan University. He was also trained in Cardiology during Postdoctoral Fellow. Now he has the expertise in gaseous molecules-mediated post-translational proteome, particular for NO-mediated S-nitrosylation in the vascular system, and also the behaviors of mitochondrial fusion/Fission that evaluating cell aging and cancer cell drug-resistance. In addition to general research interests, he also has an administrative duty as a Vice Chief of Center for Stem Cell Research of Kaohsiung Medical University.
Abstract:
Missing proteins are those genes with detectable transcriptional mRNA while missed in detecting translational proteins either by mass spectrometry or Western blot. To date, missing proteins are thought as a “KEY” in triggering many early physiological responses and then disappear after these signaling cascades are turned on. That’s why missing proteins are always “MISSED” in numerous studies. Embryo contains many definitive genes to temporarily control subsequent organogenesis. Since the decoding of human and mouse whole genome, their proteins share high similarity in amino acid sequence. The embryos taken from pregnant mice for 6.5, 7, 7.5 and 8 days, the time for the onset of neuron plate formation, were analyzed by mass spectrometry. There were 17 missing proteins identified. The Western blot showed that NKX1 (Chr#4), PTCHD1 (Chr#X), OTOGL (Chr#12) and ASXL3 (Chr#20) were increased in day 7 and 7.5, while decreased in day 8. This transient expression indicates an association of missing proteins in neuron development. As a result, the embryonic mice could be the most suitable platform for detecting missing proteins and elucidating their role in organogenesis.
Alexander I Archakov
Institute of Biomedical Chemistry, Russia
Title: Sensitivity, specificity and accuracy of the targeted (SRM) and shotgun (panoramic) mass-spectrometry technologies
Time : 14:05-14:25
Biography:
Archakov A I is a Full Member of the RAS, Professor and Scientific Advisor of Institute of Biomedical Chemistry. He had organized a scientific school to study molecular organization and functioning of oxygenase cytochrome P450-containing systems, molecular mechanisms of the structure and function of membranes and biological oxidation. Under his guidance, the institute’s members have developed a fundamentally new pharmaceutical composition “Phosphogliv” with antiviral activity for the treatment of liver diseases of various etiology. His present-day/current areas of expertise relate to research in the field of post-genomic technologies, nanobiotechnologies, proteomics, development of approaches to create personalized medicine of the future. He is the pioneer in the development of proteomics in Russia. Currently, he is the international “Human proteome” Project Coordinator in Russia/the coordinator representing Russia in the international “HP” project. He is one of the Russia’s top 100 scientists by Hirsch number-27. He is the author of more than 700 scientific works including about 482 scientific articles, 6 monographs, 30 patents and author’s certificates. He was Scientific Adviser for 15 Doctor students and more than 60 PhD theses. He is a winner of three State Prizes of the USSR, the RSFSR and of the Russian Federation, the Russian Federation Government Award, the Bach Prize of the USSR Academy, the Order "For Merit to the Fatherland” (IV, III, II class).
Abstract:
There are about 2,579 missing proteins (NeXtProt PE2,3,4) in the human proteome. They have not been detected in any biological sample using existing experimental methods. They may be not translated, or presented in low-copied state, so existing methods could not be used to detect them. In this work, we compared existing proteomic mass spectromic technologies by using UPS2 set as control (Sigma-Aldrich) and Chromosome 18 encoded proteins detected in human liver and HepG2 cells. Targeted (Selected reaction monitoring, SRM) and panoramic (Shotgun LC-MS/MS) mass spectrometric methods were compared by sensitivity, specificity and accuracy similarly to the FDA diagnostic methods. Transcriptomic analysis of the same sample of human liver and HepG2 cells, or calibration standard UPS2 set was used as a “golden” standard. Proteomic analysis results were compared with the golden standard for true and false identifications revealing. Mass spectromic methods were evaluated using FDA indicators. Sensitivity of SRM in pure UPS2 solution is 92% (44 proteins from 48 detected), and in biological matrix (E. coli extract or human blood plasma) it decreases to 63%. Shotgun LC-MS/MS reveals 23 proteins in the pure UPS2 solution and 11 proteins in E. coli extract. In HepG2 cell line and liver tissue shotgun LC-MS/MS demonstrated sensitivity 6%, and SRM - 35% with the transcriptome “golden” standard. Both methods have high specificity (more than 90%), but the accuracy is only 57% for SRM and 19% for shotgun. Using indicators of sensitivity, specificity and accuracy for proteomic methods demonstrated, that proteins are “missing” in the sample due to different reasons. For example, chemical noise from other molecules in the biological matrix may interfere signal/noise ratio. As a result, MS-signals from presenting proteins may be lost or new MS-signals may appear that is the reason of false positive results. Thus, biological matrix significantly affects the list of detected proteins, which are existing in the mixture in low-copied state (≤10-9M, 109 copies in 1Ï»L).
Recent Publications
1. Zgoda V G et al. (2013) Chromosome 18 transcriptome profiling and targeted proteome mapping in depleted plasma, liver tissue and HepG2 cells. Journal of Proteome Research. 12(1): 123–134.
2. Ponomarenko E A et al. (2014) Chromosome 18 transcriptoproteome of liver tissue and HepG2 cells and targeted proteome mapping in depleted plasma. Journal of Proteome Research. 13(1): 183–190.
3. Poverennaya E V et al. (2016) State of the art on chromosome 18-centric HPP in 2016: transcriptome and proteome profiling of liver tissue and HepG2 cells. Journal of Proteome Research. 15(11): 4030–4038.
Theo Crosson
Proteintech Group Europe, UK
Title: Fusion protein raised antibodies for evaluating mass spectrometry results
Time : 14:25-14:45
Biography:
Theo Crosson is the account manager for France territory at Proteintech. Theo has studied biology and biotechnology at the ENSTBB in Bordeaux where he got an engineering degree. He has worked in research in Next Generation sequencing for a year at New England biolabs. He then completed his education with a specialized master in biotech management at the ESC Grenoble. He is now working for Proteintech, a company committed to providing high-quality antibodies produced and validated in-house, with the aim of assisting French researchers in their use of antibodies.
Abstract:
Proteomics studies often rely on the use of antibodies. Their ability to bind any protein makes them powerful tools of use for quantification, detection, and characterisation of proteins. In particular, antibodies have been increasingly used in these past years for mass spectometry applications. Proteintech, founded by a group of scientists in 2002, is committed to manufacturing high quality antibodies in-house. Using whole-protein approach, longer time of immunization, and stringent validation procedures, Proteintech has been able to support research in proteomics. Its antibodies have been used for publications involving mass cytometry, IP-MS, and target evaluation from mass spectometry.
Wataru Aoki
Kyoto Univeristy, Japan
Title: Mixed proteome analysis to elucidate complex symbiotic interactions
Time : 14:45-15:05

Biography:
Wataru Aoki obtained his PhD degree in Biology from Kyoto University in 2013. He worked in Osaka University as Research Fellow of the Japan Society for the Promotion of Science. He is now working in Kyoto University as an Assistant Professor. His current research interests include development of technologies for next generation proteomics.
Abstract:
Proteome analysis using LC-MS/MS is an important approach for a comprehensive characterization of complex biological systems. Steady improvement in MS has transformed the depth of proteome analysis. However, the separative power of the techniques currently available is still insufficient to analyze complex proteome samples. To overcome this weak point, we have developed a monolithic super-long silica column with Dr. Minakuchi (Kyoto Monotech, http://www.k-monotech.co.jp/). The column showed very high separative performance, and LC-MS/MS system equipped with a monolithic column can identify many proteins in living cells in one-shot. Using this system, we developed a method of “mixed and quantitative proteome analysis” in which proteome samples from several different organisms were simultaneously analyzed without the need to isolate the individual living cells. Omitting the individual cell isolation steps is important because these steps are known to alter the states of protein networks by causing various artifacts from unnecessary stresses. Our methods provide novel insights into the relationship between organisms with complex interactions and should lead to a better understanding of mechanisms of infectious diseases and symbiosis.
- Bioinformatics and Computational Biology | Biostatistics - An Approach to Bioinformatics | Immunology and Drug Discovery
Location: Paris, France
Session Introduction
Dharmendra K Yadav
Gachon University, South Korea
Title: Synthesis, biological evaluation and molecular simulation studies of new arylated benzo[h]quinolines
Time : 15:05-15:25

Biography:
Dharmendra K Yadav received his PhD degree from CSIR-Central Institute of Medicinal and Aromatic Plants, Lucknow, India, in 2013. During Doctoral research, he focused on the Molecular Modeling and Drug Design. After completion of PhD, he moved to Hanyang University Seoul, Korea for Postdoctoral Research and worked on QNTR. During this period he had performed his research in Molecular Modeling on nanoparticles specially using QNTR, etc. After completion, he moved to University of Delhi, and All India Institute of Medical Sciences, Jodhpur, India. He has published more than 38 research papers in reputed international journals with high impact factor and has one US patent. He is continuing his research in Computer-Aided Drug Design Dynamics Simulation of Biological Networks and Plasma Medicine, etc.
Abstract:
In this study, we have carried out Gaussian-based 3D-QSAR model against the target COX-2 with good statistical significance (R2training=0.86) and predictability (Q2training=0.66, Q2test=0.84). The 3D-QSAR includes steric, electrostatic, hydrophobic, and hydrogen bond acceptor field indicators, whereas the potential field contributions indicate that the steric and hydrophobic features of the molecules play an important role in governing their biological activity. The anti-cancer activity of the benzo[h]quinolines was evaluated on cultured human skin cancer (G361), lung cancer (H460), breast cancer (MCF7) and colon cancer (HCT116) cell lines. The inhibitory effect of these compounds on the cell growth was determined by the MTT assay. Among the synthesized compounds 3e, 3f, 3h and 3j showed potential cytotoxicity against these human cancer cell lines. Effect of active compounds on DNA oxidation and on expression of apoptosis related gene was studied. While their bioavailability/drug-likeness was predicted to be acceptable but requires future optimization. These findings reveal that benzo[h]quinolines act as anti-cancer agents by inducing oxidative stress-mediated DNA damage. Molecular simulation study was performed to find binding conformations and different bonding behaviors, in order to reveal the possible mechanism of action behind higher accumulation of active benzo[h]quinolines with β-tubulin.
Recent Publications
1. Yadav DK, Kumar S, Saloni, Singh H, Kim MH, Sharma P. Misra S, Khan F (2017) Molecular docking, QSAR and ADMET studies of withanolide analogs against breast cancer. Drug Design, Development and Therapy 11:1859-1870.
2. Yadav DK, Rai R, Kumar N, Singh S, Misra S, Sharma P, Shaw P, Pérez-Sánchez H, Mancera RL, Choi EH, Kim MH, Pratap R (2016). New arylated benzo[h]quinolines induce anti-cancer activity by oxidative stress-mediated DNA damage. Scientific reports 6:38128.
3. Yadav DK, Dhawan S, Chauhan A, Qidwai T, Sharma P, Bhakuni RS, Dhawan OP, Khan F (2014). QSAR and docking based semi-synthesis and in vivo evaluation of artemisinin derivatives for antimalarial activity. Current Drug Target 15(8):753-61.
4. Yadav DK, Ahmad I, Shukla A, Khan F, Negi AS, Gupta A (2014). QSAR and docking studies on Chalcone derivatives for anti-tubercular activity against M. tuberculosis H37Rv. Journal of Chemometrics 28: 499-507
5. Yadav DK, Kalani K, Singh AK, Khan F, Srivastava SK, Pant AB (2014). Design, synthesis and in vitro evaluation of 18β-glycyrrhetinic Acid derivatives for anticancer activity against human breast cancer cell line MCF-7. Curr Med Chem 21(9):1160-70.
Kathy Tzeng
Optum (UnitedHealth Group), USA
Title: From genomics to personalized healthcare
Time : 15:25-15:45

Biography:
Kathy Tzeng, PhD is a Distinguished Engineer in the Advanced Applied Technology team at Optum, with a focus on Genomics and Deep Learning. Prior to joining Optum, she was IBM’s worldwide Technical Team Lead for Healthcare and Life Science in the Systems group. Before joining IBM, she worked in Boehringer Ingelheim Pharmaceuticals, where she led several drug discovery projects in the area of Bioinformatics, Proteomics and Genomics. She received her PhD in Genetics from Iowa State University.
Abstract:
With the advent of Next Generation Sequencing (NGS) technologies that have significantly reduced the cost and time required to sequence human genomes, personalized healthcare is becoming a reality. Genomic information is increasingly an important determinant of clinical diagnosis and treatment. A bioinformatics challenge central to personalized healthcare is to identify the associations between genotypic and clinical observations, and to improve treatment outcomes by associating patients with known genomic specific treatments. Several analytics approaches and reference architecture to support high velocity software and large volume of unstructured data will be discussed
Recent Publications
1. Ogasawara T, Cheng Y, Tzeng TK (2016) Sam2Bam: High Performance Framework for NGS Data Preprocessing tools. PLOS ONE. https://doi.org/10.1371/journal.pone.0167100.
2. Sachdeva V, Kistler M, Tzeng TK (2010) Enabling Bioinformatics Algorithm on the CELL BE Processor. Chapter VII in Book “Scientific Computing with Multicore and Accelerators” CRC Press.
3. Sachdeva V, Kistler M, Speight E, Tzeng TK (2008) Exploring the viability of the Cell Broadband Engine for bioinformatics applications. Parallel Computing. 34(11): 616-626.
Mari van Reenen
North-West University, South Africa
Title: XERp a novel approach to variable selection for classification
Time : 16:05-16:25

Biography:
Mari van Reenen is Head of Bioinformatics at the Centre for Human Metabolomics, North-West University (Potchefstroom Campus), South Africa as well as a PhD candidate at the Department of Statistics, Faculty of Natural Sciences North-West University (Potchefstroom Campus), South Africa. The new statistical methods presented here form part of her PhD study aimed at developing new statistical approaches that can account for the nuances of metabolomics data. Her continued work and research aims to bridge the gap between the assumptions statistical tools often require and the reality of experimental work, from experimental design to data analysis.
Abstract:
Statement of the Problem: Variable selection and classification can become complex due to the large number and sources of missing values often present in data, specifically data generated in GC-MS based metabolomics experiments. Missing values are set to zero under certain conditions resulting in a mixture distribution which are difficult for most statistical approaches to account for.
Methodology: ERp is a variable selection and classification method based on minimized classification error rates, from a control and experimental group. ERp tests the null hypothesis that there is no difference between the distributions of the two groups. Significant variables are can discriminate between the two groups and provide insight into the biological mechanisms driving group differences. XERp is an extension of ERp that takes zero-inflated data into account. XERp addresses two sources of zero-valued observations: (i) zeros reflecting the complete absence of a metabolite from a sample (true zeros); and (ii) zeros reflecting a measurement below the detection limit.
Findings: XERp performs well with regard to bias and power. XERp was also applied to a GC-MS dataset from a metabolomics study on tuberculosis meningitis in infants and children and generated a list of discriminatory variables which informed the biological interpretation of the data. XERp also attained satisfactory classification accuracy for previously unseen cases in a leave-one-out cross-validation context.
Conclusion & Significance: XERp is able to identify variables that discriminate between two groups by simultaneously extracting information from the difference in the proportion of zeros and shifts in the distributions of the non-zero observations. XERp uses simple rules to classify new subjects and a weight pair to adjust for unequal sample sizes or sensitivity and specificity requirements.
Recent Publications
1. Van Reenen, M Westerhuis J A, Reinecke C J and Venter J H (2017) Metabolomics variable selection and classification in the presence of observations below the detection limit using an extension of ERp. BMC Bioinformatics, 18(1):83.
2. Irwin C, van Reenen M, Mason S, Mienie L J, Westerhuis J A, and Reinecke C J (2016) Contribution towards a metabolite profile of the detoxification of benzoic acid through glycine conjugation: an intervention study. PloS one. 11(12): e0167309.
3. Van Reenen M Reinecke C J, Westerhuis J A and Venter J H (2016) Variable selection for binary classification using error rate p-values applied to metabolomics data. BMC Bioinformatics. 17(1):33.
4. Moutloatse G P, Bunders M J, van Reenen M, Mason S Kuijpers, T W Engelke U F and Reinecke C J (2016) Metabolic risks at birth of neonates exposed in utero to HIV-antiretroviral therapy relative to unexposed neonates: an NMR metabolomics study of cord blood. Metabolomics. 12(11):175.
5. Mason S, van Furth A M T, Solomons R, Wevers R A, van Reenen M and Reinecke C J (2016) A putative urinary biosignature for diagnosis and follow-up of tuberculous meningitis in children: outcome of a metabolomics study disclosing host–pathogen responses. Metabolomics, 12(7):1-16.
Saloni
Gachon University, South Korea
Title: Molecular dynamics simulations and ADME studies of benzopyran class of selective COX-2 inhibitors for inflammatory activity
Time : 16:25-16:45

Biography:
Saloni received her Post-graduation in Applied Chemistry from Amity University, Noida, UP, India in 2016. During her Post-graduation, she completed her summer training at the 'Centre for Aromatic Plants', Uttarakhand, India where she got brief knowledge of the different medicinal properties of various aromatic plants and also learned to use few instruments in the lab. She also worked as the Research Trainee in the Dept. of Chemistry, University of Delhi, India. She is presently working as a PhD student in College of Pharmacy, Gachon University, Incheon city, Korea. She has published two research articles in reputed international journals with high impact factor. She is continuing her research in Computer-Aided Drug Design Dynamics Simulation of Biological Networks and Plasma Medicine, etc.
Abstract:
In the present work, 3D-QSAR model was derived by partial least squares method for the prediction of anti-inflammatory activity of benzopyran class of compounds against the COX-2 (cyclooxygenase-2). Partial least squares showed high correlation significant model with (R2training=0.866) and predictability (Q2training=0.66) and indicated that physiochemical descriptors namely, steric, electrostatic, hydrophobic, and hydrogen bond acceptor field indicators, correlate well with activity, whereas the potential field contributions indicate that the steric and hydrophobic features of the molecules play an important role in governing their biological activity. A molecular docking interaction pattern analysis reveals the importance of Tyr-361 and Ser-516 of the COX-2 active site for X-ray crystal structures and this class of molecules. Thus the molecular modeling based approaches provided an improved understanding in the interaction between benzopyran class and COX-2 inhibition. These findings may be of immense importance in the anti-inflammatory drug development of an inexpensive and benzopyran class of compounds.
Recent Publications
1. Yadav DK, Kumar S, Saloni, Singh H, Kim MH, Sharma P Misra S, Khan F (2017) Molecular docking, QSAR and ADMET studies of withanolide analogs against breast cancer. Drug Design, Development and Therapy 11:1859-1870.
2. Yadav DK, Rai R, Kumar N, Singh S, Misra S, Sharma P, Shaw P, Pérez-Sánchez H, Mancera RL, Choi EH, Kim MH, Pratap R (2016) New arylated benzo[h]quinolines induce anti-cancer activity by oxidative stress-mediated DNA damage. Scientific reports 6:38128.
3. Yadav DK, Dhawan S, Chauhan A, Qidwai T, Sharma P, Bhakuni RS, Dhawan OP, Khan F (2014) QSAR and docking based semi-synthesis and in vivo evaluation of artemisinin derivatives for antimalarial activity. Current Drug Target 15(8):753-61.
4.Yadav DK, Ahmad I, Shukla A, Khan F, Negi AS, Gupta A (2014) QSAR and docking studies on Chalcone derivatives for anti-tubercular activity against M. tuberculosis H37Rv. Journal of Chemometrics 28: 499-507
5. Yadav DK, Kalani K, Singh AK, Khan F, Srivastava SK, Pant AB (2014) Design, synthesis and in vitro evaluation of 18β-glycyrrhetinic Acid derivatives for anticancer activity against human breast cancer cell line MCF-7. Curr Med Chem 21(9):1160-70.
Fatemeh Koddam
Islamic Azad University, Iran
Title: Molecular Dynamics based screening for Cag A protein: Using Quercus brantii extract substances as lead-like
Time : 16:45:17:05

Biography:
Fatemeh Khoddam is a young researcher at Azad University of Tehran. Her main research interests are bioinformatics and drug design. Fatemeh studied Biophysics (Bachelor of Science) and then complete her education in Microbiology (Master of Science) both at Azad University. After spending a term in Bioinformatics in November 2014 she started her research in a team for inhibiting Listeria disease. During her first team work experience, Fatemeh started teaching molecular biology in a private institute in Tehran and she could establish a Drug Design Department in February 2016. Fatemeh is planning to apply for PHD in Drug Design.
Abstract:
Statement of the Problem: Gastric cancer is among the fifth common malignancy and third cause of cancer related mortality worldwide (approximately 700000 victims registered each year). Helicobacter pylori, that has been closely related to gastric ulcers and adenocarcinoma, infects nearly more than 50% of the entire human population based on presence or absence of CagA gene-encoded CagA protein, H. pylori divided into 2 strains: CagA positive and CagA negative. Currently, it has been demonstrated that CagA positive strain directly effects on gastric cancer incidence, so that H. pylori infection may lead to gastric cancer if it is getting chronic. But due to antibiotics resistance module which revealed by H. pylori strains, significance of discovering new generation of antibiotics is certainly tangible.
Methodology & Theoretical: In this study Quercus brantii as an Iranian aborigine, which rich extract called Shookeh has been obtained from it, used whereas leading like compound to inhibit specifically Cag A protein. since utilization of GC MASS technique for analysis Shookeh, Furfural was identified as the most abundant compound. The structure of Cag A protein with PDB ID 4IRV used for expectancy of binding affinity between the protein and Furfural by PyRx autodock vina software. Whereof the aim of this study is to present an efficient drug-like substance we respectively engineered furfural by Hyperchem software and make carbon nano tube around substance by SAMSON software to making an improved inhibitor for Cag A protein.
Conclusion & Significance: Molecular docking analysis indicates -4.3 kcal/mol for binding affinity and pharmacokinetic analysis by using FAFDrugs4 server does not predict any oral toxicity for furfural. After engineering the substance binding affinity reduces up to -6.8 kcal/mol with no oral toxicity even with carbon nanotube.
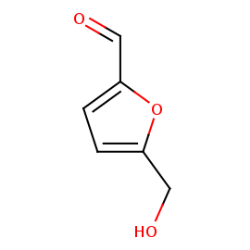
Figure 1: Furfural substance obtained from pubchem NCBI and 3D structure of Cag A protein
Recent Publications
1. HATAKEYAMA, Masanori (2017) Structure and function of Helicobacter pylori CagA, the first-identified bacterial protein involved in human cancer. Proceedings of the Japan Academy, Series B
2. Lai, Chih-Ho et al (2011) Helicobacter pyloriCagA-mediated IL-8 induction in gastric epithelial cells is cholesterol-dependent and requires the C-terminal tyrosine phosphorylation-containing domain. FEMS microbiology letters.
3. Mishra, Ajay Kumar (2013) Nanomedicine for drug delivery and therapeutics, John Wiley & Sons.
4. Shrivastava, Arpit Kumar et al (2017) Insilico identification and validation of a novel hypothetical protein in Cryptosporidium hominis and virtual screening of inhibitors as therapeutics. Parasitology research.
5. Murata-Kamiya, Naoko (2010) Helicobacter pylori exploits host membrane phosphatidylserine for delivery, localization, and pathophysiological action of the CagA oncoprotein. Cell host & microbe.
- Protein Expression and Analysis | Omics Data Integration and Databases | Mass Spectrometry in Proteome Research | Proteomics in Biochemistry and Molecular Biology
Location: Paris, France
Session Introduction
Marwa Eltoweissy
Georg August Medical University of Göttingen, Germany and Alexandria University, Egypt
Title: Short-time increase of glucose concentration in PDS results in extensive removal and high glycation level
Time : 11:20-11:40

Biography:
Marwa Eltoweissy has completed her PhD through a scholarship and cooperation work between faculty of Science, Alexandria University, Egypt and Rheinische Friedrich-Wilhelms-University Medical Center Bonn, Institute for Physiology II, Germany. She achieved Postdoctoral studies at the Gastroenterology and Endocrinology department, Georg-August University Medical Center, Göttingen, Germany. She received the Doctor of Natural Sciences degree through her work at the Nephrology and Rheumatology department, Georg-August University Medical Center, Göttingen, Germany. She worked as a major Scientific Researcher at the later department and is an Assistant Professor of Physiology at the Zoology department, Alexandria University, Egypt. She has published more than 30 papers in reputed journals and is serving as a reviewer for privileged journals. She has been involved in many international conferences and workshops as a speaker, member of Scientific Program Committees, Organizer, Session Chair/Co-chair and in conferences moderation. She is a Member of the Editorial Board of two journals in proteomics.
Abstract:
Background: Renal diseases constitute a major health risk in all societies. The prevalence of end-stage renal disease (ESRD) in adult European populations is above 10% with tendency to increase, posing a serious health threat. The treatment of ESRD involved the use of various dialysis procedures or kidney transplantation. Continuous ambulatory peritoneal dialysis (CAPD) uses the well flow peritoneum as a biological semipermeable membrane without an extra corporal blood circulation. Glucose solution is commonly used as dialysate compound. In our present study, we investigated the impact of short-time alteration of the glucose concentration and the osmolarity of the peritoneal dialysis solution (PDS) on protein removal.
Methods: Peritoneal dialysis liquids (PDL) were collected from 19 well-characterized CAPD patients treated with two types of PDS. The patients were subjected to short-time changes (4 h) of glucose concentration of PDS. The depletion of the six-interfering high abundant proteins from the PDL samples was performed with the Multiple Affinity Removal LC Column-Human 6. The resulting protein fractions were analyzed by 2D gel electrophoresis, differential in gel electrophoresis, mass spectrometry and 2D western blot.
Results: Proteomics investigation of the PDL fractions after depletion allowed the identification of 198 polypeptides, which equate to 48 non-redundant proteins. Comparative analyses of 2D gel electrophoresis protein pattern revealed a clear correlation between protein removal, PDS glucose concentration and osmolarity. An increase for 4 h in the PDS osmolarity (with 43-51 mOsmol/L) resulted qualitatively in 18-23% more protein removal in PDL. Moreover, 2D western blot analyses of the protein glycation pattern showed that the short-time increase in PDS glucose concentration (45-50 mM) resulted in significant alteration of the advanced glycosylation end products (AGEs) pattern.
Conclusions: The data presented in this study shed light on the quality of the protein lose during CAPD due to the glucose concentration in used dialysate. Moreover, we could demonstrate that higher glucose concentration in dialysis solution results in increased AGEs.
Pavlina Dolashka
Bulgarian Academy of Sciences, Bulgaria
Title: Proteomics analysis of Alzheimer's and antitumor activity of glycoproteins against bladder carcinoma
Time : 11:40-12:00

Biography:
Dolashka P and her group has wide experience in the isolation, purification, and characterization of biologically active compounds. She has more than 130 publications on these topics, 3 book chapters and 6 patents. She is Editor-in-board of 3 journals and representative IUPAC. She is coordinating several international research projects, sponsored by NATO (Brussels), the European Commission, Germany (DFG and BMBF), CNR (Italy), FWO (Belgium), China, and Ukraine.
Abstract:
Alzheimer's disease (AD) is the most common form of dementia. It is the sixth leading cause of death, and affects nearly 30 million people worldwide. Scopolamine and streptozotocin are widely utilized in chemically-induced dementia animal models to mimic specific pathophysiological pathways thought to underline AD. To the best of our knowledge, there is no report describing proteome analysis on scopolamine or streptozotocin AD animal models. Therefore, we conducted a comparative proteome analysis on CSF isolated from rats with chemically-induced dementia with the purpose of identifying protein biomarkers. Rodents were divided into three groups: rats with scopolamine-induced dementia, rats with streptozotocin-induced dementia and healthy controls. Proteins and peptides were separated from the isolated CSF into four fractions. Two low molecular peptide fractions, with mass below 3 kDa, and another with mass ranging from 3 to10 kDa were analyzed by mass spectrometry, while two other protein fractions, with mass between 10 and 50 kDa, and with mass higher than 50 kDa, were characterized by 2D-PAGE and the results were compared. The impact of hemocyanin on tumor cells was investigated by 2D-gel PAGE and several proteins showed indeed altered abundancies. The most effective inhibition of tumor cells is probably caused by a specific novel and unusual N-glycan oligosaccharide structure on HlH with methylated hexoses, an internal fucose residue connecting one GalNAc(ß1-2) and one hexuronic acid.
Theresia Conrad
Hans-Knöll-Institute, Germany
Title: Integration of multilevel OMICs data based on the identification of regulatory modules
Time : 12:00-12:20

Biography:
T Conrad studied bioinformatics at the Friedrich Schiller University Jena, Germany. During her studies, she spent several years at the Centre for Innovation Competence Septomics of the Jena University Hospital and Friedrich Schiller University focusing on the exploration of septic infections in consideration of the PIRO-concept. In 2015, she was awarded a Jena School for Microbial Communication (JSMC) fellowship and started her PhD in Bioinformatics at the Leibniz Institute for Natural Product Research and Infection Biology - Hans Knöll Institute. Her research focuses on developing multilevel models to obtain a deeper understanding of host-pathogen-interactions during fungal infections.
Abstract:
Complex scientific experiments provide researchers with a wealth of data from heterogeneous sources. Analyzed in its entirety, omics data provide a deep insight into the overall cellular processes of organisms. However, the integration of data from different cellular levels is challenging. Thus, there is a need for approaches dealing with this issue and in this study, we make use of transcriptome, proteome and secretome data from the human pathogenic fungus Aspergillus fumigatus challenged with caspofungin. Caspofungin is an antifungal drug targeting the fungal cell wall leading to a compensatory stress response. We analyze the experimental data based on two different approaches: first, we apply a simple approach based on the comparison of differentially regulated genes and proteins; secondly, we compare the cellular levels based on the identification of regulatory modules from protein-protein interaction networks. Our results show that both approaches associate the fungal caspofungin response with biological pathways like cell wall biosynthesis and carbohydrate metabolism. Compared to results of the simple approach, the regulatory modules show a notably higher consistency between the levels. The additional structural information of the networks provided by the module-based approach allows for topological analysis and the analysis of the temporal evolution of response. However, the quality of the module-based results depends on the comprehensiveness of the underlying protein-protein interaction network itself. Thus, while our results highlight the benefits and potential of a module-based analysis of multilevel omics data, future studies will have to focus on the expansion of organism specific protein-protein interaction networks.
Yinghong Pan
The National Key Facility for Crop Gene Resources and Genetic Improvement - ICS CAAS, China
Title: A proteomics strategy to analyze complex traits and gene functions
Time : 12:20-12:40

Biography:
Yinghong Pan worked for the Institute of Medicinal Plant Development (IMPLAD), Chinese Academy of Medical Sciences, and is engaged in research on medicinal plants and bioactive proteins during 1982-1999. Since 1999, he worked for the State Key Laboratory for Biology of Plant Diseases and Insect Pests (SKLBPI), Institute of Plant Protection, and the National Key Facility for Crop Gene Resources and Genetic Improvement (NFCRI), Institute of Crop Science, Chinese Academy of Agricultural Sciences, and he is engaged in research on plant proteomics. He has published more than 20 papers related to medicinal plants, and more than 55 related to proteins and proteomics.
Abstract:
Proteomics analysis of complex trait is useful to understand gene functions and trait mechanisms. But the reliability of proteomics analysis is affected by many factors, such as genetic background influences, sampling variations, and experimental conditions. In this strategy, four near isogenic lines (NILs) of dwarf male-sterile (DS) wheat with different genetic backgrounds were analyzed and a large mass spectrometry (MS) data from multiple batches were used to study the DS traits in wheat. At first, another and immature spike proteins from four NILs of DS wheat were prepared in different ways and detected with different mass spectrometry, and a total of 58170 protein groups were detected from sixteen independent experiments. Secondly the abundance distributions of proteins that detected at different frequencies and expressed types were analyzed. By using several simple formulae which were introduced to evaluate the reliability of protein expression, a database contained expression levels of 58170 protein groups and comprehensive evaluation values of 17187 proteins without duplications was established. As focusing on nuclear male sterile trait mechanism and the function of Taigu genie male-sterile wheat (TGMSW) gene ms2 in those NILs, proteomes of immature spike from three DS wheat NILs were analyzed under same conditions, and 160 differentially expressed proteins and 43 highly expressed proteins detected in this experiment were compared with database. Finally, it is determined that 28 proteins were closely related to male-sterile trait. The result show that large MS data from multiple batches is helpful for studying the complex traits and gene functions.
Leonora V Autus-Geniston
United Bayanihan Foundation and St. Paul University-Quezon City, Philippines
Title: STRING v9.1: Edges and nodes provide global functional association to protein crosstalk and integration
Time : 12:40-13:00

Biography:
Leonora V Autus-Geniston has completed his PhD from University of Santo Tomas. She is the Officer of United Bayanihan Foundation and an Assistant Professor at Saint Paul University–Quezon City, Philippines. He has published several papers in reputed journals on proteomics and pharmacogenomics, specifically on SNPs on drug metabolizing enzymes among Filipinos. She received the Best Poster Award in Health Science from National Academy of Science and Technology (Philippines) on “Method Development of nanoLC-MS/MS for Identification of Protein Biomarkers in the Urinary Proteomes of Prostate Cancer” and Best Poster in Penang, Malaysia on “Protective Effects of Thiamin on Genotoxicity in Mice”.
Abstract:
Prostate cancer was the number one cause of cancer in men in Asia. Urine like blood has a similar source of proteins making it an ideal matrix for the study. The research aims to identify proteins in the urinary proteomes of normal controls, benign prostate hyperplasia (BPH) and prostate cancer by initial separation using SDS-PAGE followed by nanoLC-Orbitrap MS/MS aided by ProteinProspector and XCalibur. STRING v9.1 predicted their functional protein association networks among the gene lists. Three hub proteins (B2M, GSN, IGHG1) and FN1 were seen in normal state and BPH, respectively. Six functional modules in normal controls and BPH were seen, and 12 in prostate cancer. Five and 15 unique proteins were seen in normal and BPH. HBA1, HBB and TTR were the distinct proteins observed in prostate cancer. A unique BPH module emerged from the normal state when their networks were overlapped. The modules at the center of the prostate cancer networks were tight and multiply connected with loosely-arranged periphery. The protein markers of prostate cancer, HBA1-HBB edge in module 1 in the periphery, while TTR in module 6 at the center was connected by haptoglobin (HP) from module 5. Prostate carcinopathogenesis from the normal state involved three protein switches: GSN-FN1, B2M-IGHG1 and CTSB-CSTB edges independent from BPH pathogenesis. From these switches, the expression of the known cancer functional modules emerged that linked further to HBA1-HBB-TTR hub. The details of these events will be discussed. Thus, the present study provides a solid framework for further understanding of prostate pathocarcinogenesis.
Altijana Hromic
University of Graz, Austria
Title: Structural view on substrate specificity of dipeptidyl peptidase III (DPP III)
Time : 14:00-14:20

Biography:
Altijana Hromic has completed her Master’s studies in Biochemistry and Molecular Biomedicine 2013 from Graz University of Technology and is attending her PhD studies at the University of Graz. She has published more than 10 papers in reputed journals and has extended experience in industry projects.
Abstract:
Dipeptidyl peptidase III (DPP III), also known as enkephalinase B, belongs to family of M49 of zinc-dependent metallopeptidases and cleaves dipeptides sequentially from the N-terminus of numerous bioactive substrates. These peptides are produced in the body and have been shown to act as neurotransmitters by interacting with their cognate receptors. Some of them form the endogenous opioid peptide system, which modulates large numbers of motivational, sensory and cognitive functions. Moreover, they contribute to the regulation of diverse physiological functions like signal transduction, gastrointestinal motility, social behavior with the aspect to drug addiction and immune functions. Many pharmacological experiments showed the role of DPP III in pain modulation and recently implicated its involvement in oxidative stress response or in the regulation of blood pressure, which fostered higher interest in investigation of this enzyme. DPP III is found in different species including higher mammals, but has also been described in lower eukaryotes like the yeast Saccharomyces cerevisiae and in some bacterial species like Porphyromonas gingivalis and Bacteroides thetaiotamicron. It has been reported that DPP III acts as post proline cleaving enzyme. This leads to the conclusion that it can develop amino acid pool by cleaving proline containing peptides which are usually resistant to hydrolysis by other aminopeptidases. This presentation provides a general overview of dipeptidyl peptidases III from different organisms, their biochemical and structural properties.
Dariusz Rakus
University of Wrocław, Poland
Title: Proteomic and immunofluorescent evidence for and against the astrocyte-to-neuron lactate shuttle
Time : 14:20-14:40

Biography:
Dariusz Rakus is the Head of the Department of Molecular Physiology and Neurobiology, at University of Wroclaw. He has published more than 50 papers in reputed journals about multifunctional proteins in glycolysis and glyconeogenesis and in the field of physiology and biochemistry of muscle, and brain, and cancer tissues.
Abstract:
Lactate derived from astrocytic glycogen has been shown to play a decisive role in induction of neuronal plasticity by stimulating memory formation in hippocampi of young animals but inhibiting it in old animals. Pharmacological inhibition of glycogen turnover in young animals blocked the basal transmission and memory formation while it improved significantly the neuronal plasticity in hippocampi of aged animals. Here we show, using quantitative proteomics and immunofluorescent microscopy, that aging is associated with an increase of glycogen metabolism enzymes concentration and shift in their localization, from astrocytes to neurons. These changes are accompanied with reorganization of hippocampal energy metabolism which is manifested by elevated capacity of aging hippocampal neurons to oxidize glucose in glycolysis and decreased ability of their mitochondria to produce energy. Our observations suggest that astrocyte-to-neuron lactate shuttle may operate in young hippocampi, however, during aging, neurons became independent on astrocytic lactate and they may start to produce lactate from blood-derived glucose.
- Bioinformatics and Computational | Bioinformatics Algorithms & Databases
Location: Paris, France
Session Introduction
K. Venkateswara Swamy
Dr. D Y Patil Biotechnology and Bioinformatics Institute, India
Title: Molecular modeling, docking, dynamics and simulation of deguelin and its derivatives with cyclin D1 and cyclin E in cancer cell signaling pathway
Time : 14:40-15:00

Biography:
K Venkateswara Swamy is currently working as an Associate Professor. He was awarded Young Scientist Award by ICMPHP, George Washington University, USA in 2013. He is a Visiting Faculty at Jaffna University, Sri Lanka. He has received SERB-DST project under Young Scientist Scheme in the year 2016 and is a member in various academic committee of bioinformatics at Dr. D Y Patil Vidyapeeth, Pune, India. He has published 23 national and international research papers in reputed journals. He was invited for national workshop in bioinformatics as Key Note Speaker by various Universities and Institutes. He is expertise with Molecular Modeling, Docking, Simulations and Dynamics of cancer proteins. He has already submitted 32 theoretical protein models to Protein Model Data Base (PMDB).
Abstract:
Deguelin is a major active ingredient and principal component in several plants, Derris trifoliata Lour. (Leguminosae), Mundulea sericea (Leguminosae), Tephrosia vogelii Hook.f. (Leguminosae) and potential molecule to target cancer cell signaling pathway proteins. As a complex natural extract, deguelin interacts with various molecular targets to exert its anti-tumor properties at nanomolar levels. Deguelin induced cell apoptosis by blocking anti-apoptotic pathways, while inhibiting tumor cell propagation and malignant transformation through p27-cyclin-E-pRb-E2F1- cell cycle control and HIF-1αVEGF antiangiogenic pathways. Our research explores the deguelin and its derivatives interaction with crystal structure of cyclin D1 (PDB ID: 2W96) and cyclin E (PDB ID: 2AST) to understand the better molecular insights. Molecular modelling of ligands (deguelin and its derivatives) were carried out by Avogadro software till stable confirmation obtained. The partial charges for the ligands were assigned as per standard protocol for molecular docking. All docking simulation were performed with AutoDock Vina on workstation. Virtual screening was done for all docked molecules based on binding energy and hydrogen bonding affinity. Molecular dynamics (MD) and Simulation (10 ns and 12 ns for cyclin D1 and cyclin E1 respectively) was done using GROMACS 5.1.1 software to explore the interaction stability. All the stable confirmations for cyclin D1 and cyclin E proteins trajectories was captured at various time intervals. Few compounds screened based on high affinity as inhibitors for cyclin D1 and cyclin E and may inhibit the cell cycle in cancer cell signaling under in vitro and in vivo experiments.
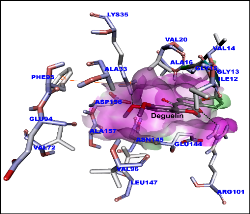
Figure 1: Molecular docking of deguelin with cyclin D1 protein. Surface shown in pink. Amino acids and deguelin hydrogen bond interaction with dotted lines.
Recent Publications
1. Prasad Dandawate, Kiranmayi Vemuri, K Venkateswara Swamy, Ejazuddin M Khan, Manjula Sritharan and Subhash Padhye (2014) Synthesis, characterization, molecular docking and anti-tubercular activity of Plumbagin-Isoniazid Analog and its β-cyclodextrin conjugate. Bioorganic & Medicinal Chemistry Letters 24(21):5070-5075.
2. Prasad Dandawate, Aamir Ahmad, Jyoti Deshpande, K Venkateswara Swamy, Ejazuddin M Khan, Madhukar Khetmalas, Subhash Padhye and Fazlul Sarkar (2014) Anticancer phytochemical analogs 37: Synthesis, characterization, molecular docking and cytotoxicity of novel plumbagin hydrazones against breast cancer cells. Bioorganic & Medicinal Chemistry Letters, 24:2900-2904.
3. P Sharma, P Patil, N Rao, K V Swamy, M B Khetmalas and G D Tandon (2014) Mapping biodiversity of indigenous freshwater chlorophytes. Research Journal of Pharmaceutical, Biological and Chemical Sciences 5(3):1632-1639.
4. Shibnath Ghataka, Alok Vyas, Suniti Misra, Paul O’Briend, Ajit Zambre, Victor M Fresco, Roger R Markwald, K Venkateshwara Swamy, Zahra Afrasiabif, Amitava Choudhury, Madhukar Khetmalas and Subhash Padhye (2013) Novel di-tertiary-butyl phenylhydrazones as dual cyclooxygenase-2/5-lipoxygenase inhibitors: synthesis, COX/LOX inhibition, molecular modeling, and insights into their cytotoxicities. Bioorganic & Medicinal Chemistry Letters 24:317-324.
5. Siddiqui A, Dandawate P, Rub R, Padhye S, Aphale S, Moghe A, Jagyasi A, Venkateswara Swamy K, Singh B, Chatterjee A, Ronghe A and Bhat H K (2013) Novel Aza-resveratrol analogs: Synthesis, characterization and anticancer activity against breast cancer cell lines. Bioorganic & Medicinal Chemistry Letters 23:635–640.
Sherief El Rweney
Royal Holloway, University of London
Title: Drug repositioning system using the power of network analysis and machine learning to predict new indications for the approved drugs “drug repositioning and rate the level of the drug similarityâ€
Time : 15:00-15:20

Biography:
Sherief El Rweney has over 13 years experience in computer science systems and information technology, he has done Master’s degree at Royal Holloway University of London in Computer Science, Data Analysis, Machine Learning, And Bioinformatics , the merging between his experience in bioinformatics and machine learning study motivated him to build the Drug Repositioning System based on protein interaction where can be used to predict new indication for the approved drugs and help drugs scientists to go easy through their drug discovery research. He is aiming to develop the Drug Repositioning System by merging many other factors beside the protein interaction to strength the predication of the drugs repositioning.
Abstract:
Drug discovery is a lengthy process, taking on average 12 years for the drugs to reach the market –but as Sir James Black OM once said, “the best way to discover a new drug is to start with the old one”. As result, this will drive to Drug repositioning concept. Drug Repurposing and repositioning is finding a new clinical use for an approved drug. There are many factors that can be used to predict new target disease. i.e. protein-protein interaction, chemical structure, gene expression and functional genomics, Phenotype and side effect, genetic variation and Machine learning. Protein-protein interaction (PPI) is Physical contacts with molecular docking between proteins that occur in a cell or in a living organism in vivo. There are Two Alternative Approaches PPI “Binary: yeast twoâ€hybrid (Y2H) and coâ€complex: (TAPâ€MS)”. Drug Repositioning System, is a system built based on protein-protein Binary interaction to predict new targets for the approved drugs. The system curates the data sets for human PPI, Drugs and diseases from well-known online sources (PPI from HRPD, drugs from DrugBank, Diseases from DisGeNET), Drug Repositioning System relates the 3 data sets based on genes name. Drug Repositioning System consisting of two interfaces: backend system where the curated data sets stored based on rational database and frontend web interface where the end users can use many search engines to search inside the system for diseases, genes and drugs to predict and find new targets for the approved drugs based on protein interactions, from the web interface the user can make analysis based on his search result and build network between the genes, diseases and drugs and generate statistics to be able to answer his question. There are many Questions that can be answered by Drug Repositioning System and generate statistics: for example the main question is can we find new indications for existing approved drugs. Drug similarity: from the Drug Repositioning System we can measure the percentage of drugs similarity between any pair genes interaction based on the number of shared drugs between them to rate the level of drug repositioning strength and then use the ROC ( receiver operating characteristic curve) analysis.
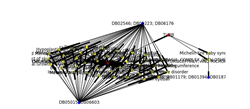
Figure 2: in the left side of the picture, it’s clear that there are two groups of drugs target numerous diseases related to HDAC6 gene, and also on the right side we will find one group of drugs targets two groups of disease that are related to TUBB gene while there link between TUBB and HDAC6 indicates the interaction between them. As result we can make drug repositioning between the two genes.
- Poster Presentations
Location: Paris, France
Session Introduction
Mohammed Sulaiman Al-Hajou
King Abdulaziz University, Saudi Arabia
Title: Impact of sequential passage on overall protein expression using proteomic analysis
Biography:
Mohammed Al-Hajouj, Master Student – Clinical Chemistry, Clinical Laboratory Science College of Applied Medical Science, King Abdulaziz University.
Abstract:
Urinary tract infection (UTI) is considered to be one of the most prevalent bacterial infections in the world predominantly affecting the bladder and the kidney; according to research, one in three women is affected by UTIs. Gram-negative bacteria are a major cause of such infections, particularly Escherichia coli (E. coli). E. coli was the main causative agent of 80-90% of community-acquired infection, about 40% of nosocomial UTI, and is responsible for 25% of recurrent infections. The field of proteomics has emerged as a great tool to analyze expressed proteins and to identify possible biomarkers associated with a number of pathological states, and to the same extent associated with bacterial pathogenesis. Researchers elsewhere are investigating E. coli proteomics profiles to identify possible biomarkers, however, protein profiles could vary environmental stress created by subculturing. Here we propose to answer the research questions, are there differences in protein profiles of E. coli originated from a sequential passage? To conduct this research, urine samples will be collected from individuals with recurrent UTI, sequentially subcultured, and analyzed using two-dimensional gel electrophoresis and mass spectrometry to identify any significant change in the protein profile of the bacteria. We hope to elucidate to the effect of passage on the protein profile of this common pathogen.
Maryam Bahmanzadeh
Hamadan University of Medical Sciences, Iran
Title: A proteomic analysis of human follicular fluid: Novel candidate markers for oocyte number and reproductive aging
Biography:
Maryam Bahmanzadeh has completed her PhD from Ahvaz Jundishapur University of Medical Sciences, Ahvaz, Iran. She is an Assistant Professor in Anatomical Sciences
Hamadan University of Medical Sciences, Iran and a Clinical Embryologist. She has published more than 10 papers in reputed journals.
Abstract:
Follicular fluid (FF) results from the transfer of blood plasma components and the secretory activity of the oocyte, granulosa and thecal cells. Certain components of FF might be used as indicators for the maturation and the quantity of the oocytes. Proteins can be used as biomarkers for reproductive diseases using both FF and plasma. Age-related infertility is usually considered as a problem that can be solved by assisted reproduction technology. Therefore, the identiï¬cation of novel biomarkers that are linked to reproductive aging is the subject of this study. FF was obtained from healthy younger (20–32 years old) and older (38–42 years old) women undergoing intracytoplasmic sperm injection (ICSI) due to male factor infertility. In this study, we investigated the protein composition of human FF obtained from females undergoing ICSI using the matrix-assisted laser desorption-ionization time-of-flight/time-of-flight (MALDI-TOF-TOF) mass spectrometry technique. Twenty-three protein spots showed reproducible and significant changes in the aged compared to the young group. Of these, 19 protein spots could be identified using MALDI-TOF-TOF-MS. As a result of MASCOT search, five unique downregulated proteins were identified in the older group. These were identified as serotransferrin, hemopexin precursor, complement C3, C4 and kininogen. A number of protein markers were found that may help develop diagnostic methods of infertility.
Qurat-ul-Ain
Dr. Panjwani Center for Molecular Medicine and Drug Research - ICCBS, Pakistan
Title: Modulation of melanoma cell proliferation and spreading by novel small molecular weight bioactive compound
Biography:
Qurat-ul-Ain, is a Ph.D student at Dr.Panjwani Center for Molecular Medicine and Drug Research, International Center for Chemical and Biological Sciences, University of Karachi. She is collaborative Research Student at Department of Dermatology and Allergy University of Ulm Germany, Ms. Qurat-ul-Ain has characterized a variety of newly designed chemical compounds in term of their antioxidant and pro-oxidant properties. She studies these in melanoma cell and melanocytes. Some of the studied compounds reveal an inhibitory effect on melanoma cells, but not on their benign counterpart, the melanocyte. The mechanisms of action of these compounds are being investigated.Qurat-ul-Ain has authored 19 articles published in international journals.
Abstract:
Melanoma, the most dangerous skin cancer, originates from the melanocytes and has a high tendency to invade neighboring tissues, and metastasize. Both bioactive compounds appear to be involved in the modulation of melanocyte transformation and melanoma progression as well as invasion. Consequently, potent bioactive compounds may prevent transformation and progression of the tumor. Melanoma is also the most common malignancy diagnosed in United States. Out of 3.5 million, 2 million cancer cases diagnosed in US annually, resulting mostly in-patient death and poor survival. With emerging new therapies early detection and treatment has resulted in high survival rates with 85% of patients surviving for at least 10 years. One of these upcoming therapeutics is the use of bioactive compounds, which have proven to show increase response rate (IRR) and overall survival (OS) rates. In fact, dacarbazine is the only FDA approved chemotherapeutic bioactive compound for melanoma treatment. Therefore, we have identified eight bioactive compounds of four different new synthetic classes synthesized by our collaborator as anti-melanoma/anti invasion agents. These compounds were previously found to posse’s antioxidant and prooxidant in an in vitro antioxidant assay. Compounds 1, 2, 3, 4, 5, 6, 7 and 8 were found to be most potent anti-melanoma agent. We are now testing these compounds for their migration inhibition role in skin melanoma as well as skin stem cells in vitro and their ability to reduce proliferation and spreading. We are currently analyzing the influence of these compounds on the molecular pathways, involved in the proliferation-invasion inhibition.
Recent Publication
- Barakat A, Ghabbour H A, Al-Majid A M, Imad Qurat-ul-Ain R, Javaid K, Shaikh N N and Wadood A (2016) Synthesis, X-ray crystal structures, biological evaluation, and molecular docking studies of a series of barbiturate derivatives. Journal of Chemistry 1-11.
- Donat-Vargas C, Berglund M, Glynn A, Wolk A and Åkesson A (2017) Dietary polychlorinated biphenyls, long-chain n-3 polyunsaturated fatty acids and incidence of malignant melanoma. European Journal of Cancer 72:137-143.
Pratibha Sharma
All India Institute of Medical Sciences, India
Title: Hippocampal neural proteins interaction of Alzheimer’s peptide: 14-3-3 zeta as potential therapeutic biomarker
Biography:
Abstract:
Introduction: Alzheimer’s disease (AD) is the most common form of dementia. Amyloid beta(1-42) is prone to aggregate and found in plaque interacting with various proteins. Low molecular weight oligomeric form of amyloid beta is found as most toxic and responsible for disease process. Hippocampus is the primary region of the brain affected by AD. In this study, interaction profiles of oligomer and monomer form of amyloid-beta binding hippocampal neuron intracellular and cytosolic proteins were obtained.
Methods: Amyloid beta (1-42) peptide- monomer and oligomer forms separated using gel-filtration chromatography. These were allowed to pull-down separately with hippocampal tissue neuron plasma membrane and cytosolic proteins using affinity chromatography in proteins native form. Interacting proteins were digested in-solution by trypsin and identified using mass spectrometry ESI-Triple-TOF5600, searched Uniprot database using software Protein pilot 4.2. Gene ontology and pathway analysis software Cytoscape was used for classification and interactions of proteins with respect to AD and apoptosis pathways.
Result: Proteins found unique binding amyloid beta peptide monomer are synuclein-beta, SNAP25, lipophilin, EAAT-2 and SRPRB. Proteins found unique binding amyloid beta peptide oligomer are tubulin-beta, Tau (MAPT), 14-3-3 and phospholipase A2.
Conclusion: Oligomer binding proteins may help in understanding the disease toxicity mechanism. Whereas, 14-3-3 protein could be possible novel therapeutic target for diagnosis, treatment and in understanding progression of AD.
Biography:
Enass Y Osman is a Lecturer of Pharmacology and Toxicology, Faculty of Pharmacy, Tanta University, Egypt. Her expertise lies in pharmacotherapy and improvement of patients’ compliances especially those with psychiatric disorders. My publications are directed toward introduction of new drugs for treatment of schizophrenic patients for improving their life. The paper is based on previous publications by Abekawa et. al. (2008) who used amphetamine for induction of schizophrenia in animals. Our researches aimed to investigate potential effects of drugs other than classical antipsychotics as adjuvant therapies in schizophrenia.
Abstract:
Schizophrenia is a complex psychiatric disorder which markedly diminishes quality of life by its effects on cognitive, behavioral and emotional areas of functioning. The exact mechanism by which schizophrenia evolves is still unknown. Genetic, environmental factors, neurotransmitter, inflammation and oxidative stress are involved in pathophysiology of schizophrenia. β-glucuronidase is a member of the lysosomal glycosidase family that catalyzes breakdown of complex carbohydrates (hydrolysis of β-D-glucuronic acid residues) from the non-reducing end of mucopolysaccharides. The present study aimed to investigate the possible effects of celecoxib and omega-3 fatty acids on inflammation and lysosomal integrity as pathophysiological markers of schizophrenia. In the present study, amphetamine-treated rats received either ripseridone, celecoxib, omega-3 fatty acids, ripseridone plus celecoxib or ripseridone plus omega-3. The effects of treatment on brain β-glucuronidase enzyme activity and inflammatory markers including IL-6, Cox-2 and NF-kB were evaluated. Treatment with celecoxib or omega-3 fatty acids alone significantly reduced neurotoxicity which was indicated by reduction of brain β-glucuronidase enzyme activity. The anti-inflammatory effects of celecoxib and omega3 fatty acids were indicted by reduction of brain IL-6 level and decreased expression of brain Cox-2 and NF-kB. Addition of celecoxib or omega-3 fatty acids to risperidone also potentiated its effects on the measured parameters. In conclusion, celecoxib and omega-3 may be promising candidates as adjuvant therapy with risperidone to enhance its outcomes in schizophrenia.
Recent Publications
1. Shakoor S, McGuire P, Cardno AG, Plomin R, Ronald A (2015) A shared genetic propensity underlies experiences of bullying victimization in late childhood and self-rated paranoid thinking in adolescence. Schizophr. Bull. 41(3):754-763.
2. El Sisi A E, Sokkar S S, ElSayad M E, Ramadan E S, Osman E Y (2016) Celecoxib and omega-3 fatty acids alone and in combination with risperidone affect the behavior and brain biochemistry in amphetamine-induced model of schizophrenia. Biomedicine & Pharmacotherapy. 82:425-431.
3. Goddard A, Leisewitz A, Kjelgaard-Hansen M, Annemarie T et. al. (2016) Excessive pro-inflammatory serum cytokine concentrations in virulent canine babesiosis. PLoS ONE. 11(3):e0150113.
4. Messamore E and McNamara R K (2016) Detection and treatment of omega-3 fatty acid deficiency in psychiatric practice: Rationale and implementation. Lipids Health Dis. 15:25.
5. Ettinger U, Meyhöfer I, Steffens M, Wagner M, Koutsouleris N (2014) Genetics, cognition, and neurobiology of schizotypal personality: a review of the overlap with schizophrenia. Front. Psychiatry 5:18.
Gauri Garg Dhingra
University of Delhi, India
Title: Genome analysis of Sphingopyxis flava R11HT: An overview of genetic attributes
Biography:
Dhingra G is an Assistant Professor of Zoology at the University of Delhi, Kirori Mal College - Department of Zoology, and has been involved in undergraduate teaching since past 13 years. She completed her PhD under supervision of Prof. Rup Lal, Department of Zoology in 2005 on Manipulations of Rifamycin Biosynthetic Gene Clusters in Amycolatopsis mediterranei. Presently she is working with Rup Lal’s group focusing on metagenomic diversity of pesticide contaminated sites.
Abstract:
A HCH tolerant strain, R11HT was isolated from the soil sample of HCH dumpsite located at Ummari village, Lucknow, Uttar Pradesh, India. On the basis of 16S rRNA gene similarity, the strain was identified as the member of genus Sphingopyxis with highest identity with Sphingopyxis indica DS15T (97.85 %). In addition, three more strains were identified with similarity >97%. The pairwise DDH analysis revealed that the strain R11HT belongs to a separate species. Further biochemical and chemotaxonomic studies confirmed the novelty of the strain and thus designated as Sphingopyxis flava R11HT. Due to the HCH tolerance ability, strain R11HT was selected for genome sequencing using Illumina Hiseq technology. It has a genome size of 4.15 Mbp, with G+C content of 63.75% and 90.40% coding potential. The strain was found to code for 4185 protein coding sequences. The total number of RNA coded by the strain is equal to 58 with a single copy of 5S, 16S and 23S rRNA and 46 tRNAs. The pfam analysis revealed 3219 protein sequences engaged in variety of metabolic functions, dominated by amino acid transport and metabolism, energy production and conversion, lipid transport and metabolism, replication, transcription and translation. The strain was also found to harbor five biosynthetic gene clusters including terpene and ectoine. The KEGG pathway genes include degradation pathways for number of compounds including amino-benzoate, benzoate, bisphenol, caprolactlam, chloroalkene, chlorohexane but lin genes which are known for HCH degradation were not annotated in the genome. It will be interesting to further analyze the probable metabolic pathways of strain R11HT which helps the strain to withstand in such high concentration of HCH.
Recent Publications
1. Verma H, Bajaj A, Kumar R, Kaur J, Anand S, Nayyar N, Puri A, Singh Y, Khurana JP, Lal R (2017) Genome organization of Sphingobium indicum B90A: an archetypal hexachlorocyclohexane (HCH) degrading genotype. Genome Biol Evol. DOI: 1093/gbe/evx133.
2. Mahato NK, Gupta V, Singh P, Kumari R, Verma H, Tripathi C, Rani P, Sharma A, Singhvi N, Sood U, Hira P, Kohli P, Nayyar N, Puri A, Bajaj A, Kumar R, Negi V, Talwar C, Khurana H, Nagar S, Sharma M, Mishra H, Singh AK, Dhingra G, Negi RK, Shakarad M, Singh Y, Lal R (2017) Microbial taxonomy in the era of OMICS: application of DNA sequences, computational tools and techniques. Antonie Van Leeuwenhoek. doi: 10.1007/s10482-017-0928-1.
3. Kumar R, Verma H, Haider S, Bajaj A, Sood U, Ponnusamy K, Nagar S, Shakarad M, Negi R, Singh Y, Khurana J, Gilbert J, Lal R (2017) Comparative genomic analysis reveals habitat specific genes and regulatory hubs within the genus Novosphingobium. mSystems. 10.1128/mSystems.00020-17.
4. Verma H, Rani P, Singh AK, Kumar R, Dwivedi V and Lal R (2015) Sphingopyxis flava sp. nov., isolated from an hexachlorocyclohexane (HCH) contaminated soil, India. Int. J. Syst. Evol. Microbiol. 65: 3720-3726.
5. Verma H, Kumar R, Oldach P, Sangwan N, Khurana J P, Gilbert J A, Lal R (2014) Comparative genomic analysis of nine Sphingobium strains: Insights into their evolution and Hexachlorocyclohexane (HCH) degradation pathway. BMC Genomics, 15:1014.
Seo Hyein
Korean Advanced Institute of Science and Technology, South Korea
Title: Development of duplicated-indel based biomarker for AML diagnosis
Biography:
Seo Hyein received the BS and MS degrees from the School of Electrical Engineering (EE), KNU, Daegu, Korea in 2014 and KAIST, Daejeon, Korea in 2016. Her research interests include WLAN, sensor network and bioinformatics. She is now a Ph.D. student of the School of EE, KAIST.
Abstract:
Statement of the Problem: Since the development of the Next Generation Sequencing (NGS), mutations in whole genome of human are investigated and the relationship between mutations and diseases, especially cancers, is studied. Insertion and deletion (Indel) is one of the important mutations. However, analysis result about the relationship between indel and cancer using NGS data is not sufficient. In this research, we want to derive the indel based biomarker that can distinguish whether the sequence is extracted from the cancer cell or not. Among indels, we use the duplicated-indel, which has similar property to microsatellite and exits in gene.
Methodology & Theoretical Orientation: We obtained mutations from all genes of 100 sequences using GATK tool. Sequences were provided by the TCGA project and were sequenced from the cancer cell and the normal cell for 50 Acute Myeloid Leukemia (AML) patients. Among mutations, duplicated-indels, which are frequently found at the cancer cell but not in the normal cell, were listed. By counting the number of listed duplicated-indels, we could generate the biomarker of the cancer.
Findings: In Fig. 1, we used 50 frequently existing duplicated-indels for the cancer distinction. By selecting the diagnosis threshold as 9 (Th1), we could accurately find 12 cancer sequences. Otherwise, if the threshold was 5 (Th2), 38 sequences were rightly extracted as the cancer but there was one wrong diagnosis.
Conclusion & Significance: We showed the probability of the cancer biomarker using the duplicated-indel from NGS data. With optimization of threshold, more accurate biomarker can be derived. Also, we can extend our research to the analysis of the relationship between microsatellite and diseases.
Recent Publications
1.Sun Q Y, et al. (2017) Ordering of mutations in acute myeloid leukemia with partial tandem duplication of MLL (MLL-PTD). Leukemia 31(1): 1-10.
2. Papaemmanuil E, et al. (2016) Genomic classification and prognosis in acute myeloid leukemia. New England Journal of Medicine 374(23): 2209-2221.
3. Rabbani B, Nakaoka H, Akhondzadeh S, Tekin M and Mahdieh N (2016) Next generation sequencing: implications in personalized medicine and pharmacogenomics. Molecular BioSystems 12(6): 1818-1830.
4. Carethers J M and Jung B H (2015) Genetics and genetic biomarkers in sporadic colorectal cancer. Gastroenterology 149(5): 1177-1190.
5. Ahmed D, et al. (2013) Epigenetic and genetic features of 24 colon cancer cell lines. Oncogenesis 2(9): e71.
Yuko Sakajiri
Iwate University, Japan
Title: Ser 81 of Natronomonas pharaonis halorhodopsin plays a role of holding chloride ions near the Schiff base
Biography:
Yuko Sakajiri has her expertise in bioinformatics of protein structure prediction and has passion in visual restore. In a previous study, she studied the protein folding prediction of the beta barrel protein and found several folding patterns in the early stages of protein folding using the average distance map technique. Then, she starts the study for visual restore and has predicted the structure of the rhodopsin-like protein and the ion pathway inside the protein. These studies are expected to lead to the development of modified rhodopsin for the improvement of visual restore.
Abstract:
In this study, we aim for advanced visual restoration of enhanced contrast by light-driven halorhodopsin into retinal photoreceptor cells and hyperpolarization of optic nerve by light absorption. However, strong light is necessary for the light driving, because halorhodopsin has low photoresponsiveness. Therefore, it is necessary to develop a functionally enhanced halorhodopsin for visual restoration. As basic research, we analyzed the amino acid residues essential for chloride ion pump of halorhodopsin.
Natronobacterium pharaonis halorhodopsin (NpHR) is a light-driven chloride ion pump. It is necessary to load chloride ions at the binding site-1 (BS1) of Thr126 near the protonated Schiff base in order to make chloride ions to pass through the cell membrane at light absorption. However, the network of hydrogen bonds near the BS1 is complicatedly intertwined, and it is not known well yet how chloride ions are retained at the BS1. In this study, we performed a molecular dynamics simulation for wild type and S81A mutant of NpHR structures, respectively to obtain dynamic information of chloride ion at the BS1. As a result, the Thr126 of the wild type retained binding to the chloride ion by hydrogen bond. While, the S81A mutant was unable for chloride ion to retain the position of the BS1, and then the chloride ion was released from there. We found that the side chain of Thr126, which was fixed by hydroxyl group of the Ser81, rotated other direction. Furthermore, NpHR S81A recombinant cell was actually prepared and then was recorded by the patch clamp. It was confirmed that the NpHR recombinant cell did not occurred hyperpolarization, and thus it was found that the NpHR S81A did not transport the chloride ion toward cytoplasmic side.
Recent Publications
1. Analysis of the Local Sequences of Folding Sites in beta Sandwich Proteins with Inter-Residue Average Distance Statistics. The Open Bioinformatics Journal 2011, 5:59-68 Yuko Ishizuka and Takeshi Kikuchi
Sotiris Avgousti
Cyprus University of Technology, Cyprus
Title: Clinical trials and evaluation of cardiac ultrasonography over 4G wireless network using a teleoperated robot
Biography:
Sotiris Avgousti, is currently working as an instructor at Cyprus University of Technology. He has done masters in Computer Systems Networking and Telecommunication from Brunel University, London. He has done PhD from the University of Oreland France in Telerobotics.
Abstract:
A wearable tele-echography robot (MELODY) with four degrees-of-freedom that permits a medical expert to examine at a distance a patient by ultrasound was evaluated and tested over 4G mobile networks. At the expert side, the medical expert uses a dummy probe to control the real probe which is controlled by the robotic arms at the patient side and positioned on the patient’s body by paramedic personnel. The communication between the two sites is facilitated by a videoconferencing link. The telerobotic system has now been clinically tested and commercialized. The experimental setup of a wearable tele-echography robot, the MELODY system over a 4G connectivity link used to measure the system performance is described. The evaluation and investigation of the relevant medical ultrasound video and the relevant issues defined in terms of the average throughput, and jitter delay are investigated. A comprehensive video coding standards comparison for cardiac ultrasound applications is performed, including H.264/AVC and HEVC using a data set of nine cardiac ultrasound videos. Both objective and subjective (clinical) video quality assessment were performed.
Biography:
Pushpanjali Dasauni is pursuing her PhD from University of Delhi South Campus, New Delhi, India and will submit the same by this year-end. She is expert in most of the proteomics tools like MALDI TOF-TOF and 2D-DIGE and works with various biophysical tools like FTIR etc. She is developing novel diagnostics tools for fast, cost-effective and accurate detection of hemoglobin disorders. She has published papers in reputed journals and she is a Life Time Member of Protein Society, India.
Abstract:
Around 7% of the global population carries an abnormal hemoglobin gene. Over 330,000 infants are born annually with hemoglobinopathies and it is the major cause of morbidity and mortality in early childhood. The treatments rely heavily on the diagnosis of hemoglobin variants. The routine/conventional techniques used for the identification of mutation in hemoglobin variants have their own limitations like co-migration of variants in electrophoresis and co-elution in HPLC. The WHO (2002) report on Genomics and Health has emphasized on the development of precise molecular techniques for screening of hemoglobin disorders. A sensitive, robust and reproducible method was thus developed to identify single substitution mutations in the hemoglobin disorders from sequence of the entire globin chains. The method was MS compatible and dealt with certain limitations like difficulty in getting complete sequence coverage. Different methods like using organic solvent, digestion with a different protease and combining results, treating the digestion mixture with 10% acetonitrile prior to incubation and combining the separation power of LC coupled with MALDI MS/MS were tried for standardization and optimization of protocol. Finally, we optimized a method using an organic solvent and heat denaturation step prior to digestion resulting in 100% sequence coverage in the β chains and 95% sequence coverage in the α chains. A hemoglobin variant database was created to specify the search and reduce the search time. All the mutations were thus identified using a non-targeted approach and this method could be used in future for regular screening of any single mutation in hemoglobin variants.
Priscilla Nascimento Guasti
Sao Paulo State University, Brazil
Title: Seminal plasma proteins in castrated stallions: partial results
Biography:
Guasti P N has a DVM from Federal Rural University of Rio de Janeiro. She obtained her Master’s Degree and PhD in Animal Reproduction from Sao Paulo State University (UNESP/Botucatu). She is currently Postdoctoral Researcher at the Sao Paulo State University. Her research interest focuses on the proteomics of equine seminal plasma, sperm membrane and epididymal fluid and its relations with the fertility.
Abstract:
Seminal plasma (SP) consists of secretions produced by testes, epididymides and accessory sex glands (AG); and contains several compounds that play important roles in sperm maturation and fertilization. The aim of this study was to investigate the expression of SP proteins after orchiectomy in stallions. Fourteen stallions were submitted to semen collection and orchiectomy. Fifteen and 30 days after the procedure, semen collection was performed again. SP samples were centrifuged and determination of total protein was performed by BCA-assay. SP proteins before (SP) and after castration (D15; D30) were analyzed by 2D-LC-MS/MS. Different proteins were identified among groups; 15 proteins in SP, 13 in D15 and 15 in D30. Castration resulted in a decrease in proteins related to reproductive process. Four proteins (KLK1E2, ALB, CLU, Equ-c1) were common to all groups and were involved in binding and catalytic activity. Catalase was identified only in castrated animals; nine proteins were identified only in D30, which were involved in immune system and metabolic process, angiogenesis and biological adhesion. In general, AG conforms to the general pattern of steroid-regulated systems as they are under androgenic control, in which hormone withdrawal may lead to differential decreases in protein expression. SP protein expression is not immediately terminated after castration, possibly due to residual levels of testosterone (under analysis). In conclusion, removal of testes influenced the expression of SP proteins and revealed exclusively expressed proteins related to the inflammatory response and tissue reconstruction; the proteins involved in reproductive process were underexpressed possibly due to decreased testosterone.
Sung Han Kim
National Cancer Center, South Korea
Title: ESRP1 has a prognostic value in biochemical recurrence and cancer specific survival of prostate cancer
Biography:
Sung Han Kim has completed his MD from Seoul National University of Medicine, Seoul, Korea and Postdoctoral studies from National Cancer Center, Goyang, Korea and Seoul National University of Medicine, Seoul , Korea. He is the Clinical Staff and Associate Researcher and Director of Jinsoo Chung, Prostate Cancer Center, National Cancer Center, Goyang, Korea. He has published more than 30 papers in reputed journals.
Abstract:
The study was aimed to identify targetable genes in PC from The Cancer Genome Atlas (TCGA), and to validate the significance of the genes identified in clinical studies using immunohistochemistry in PC patients’ prostatectomy microarray. Omics data and clinical data of 550 PC patients were obtained from TCGA. Several significant genes were identified from TCGA dataset having the most number of point mutation to exhibit significantly strong positive relationship in expression values with the most frequently mutated gene. Further validation of different expression values for the list of genes between tumor and normal lesions was performed to evaluate their prognostic significance using tissue microarray of 514 prostatectomy specimens by performing immunohistochemistry in clinical setting from a single cancer institution. Prognostic powers of these genes were investigated on the NCC dataset. Immunohistochemistry were performed on the genes associated with the most mutated gene. The gene markers’ prognostic factors were analyzed using Cox proportional hazard analysis with a significant p-value<0.05. FRG1B gene was found lineage-specific mutation with ESRP1, RAD51, and CHEK2 genes in the TCGA dataset. The union of samples with these three up-regulated markers with FRG1B mutation showed significant differences in disease-free survival compared to the samples without expression of two combinational series (p<0.05). Analysis of the clinicopathological factors related to three markers suggested that expression of ESRP1 was a significant risk factor for biochemical recurrence (BCR, HR 1.003) and cancer-specific survival (HR 1.048), even after adjusting for significant prognostic clinicopathological factors of BCR and CSS (p<0.05). On the other hand, CHEK2 was not significant in any BCR and CSS. RAD51 could not be evaluated because of its overall overexpressed in specimens. The study identified that ESRP1 was a potentially significant target gene of survival prognoses in PC.
Biography:
Sung Han Kim has completed his MD from Seoul National University of Medicine, Seoul, Korea and Postdoctoral studies from National Cancer Center, Goyang, Korea and Seoul National University of Medicine, Seoul, Korea. He is the Clinical Staff and Associate Researcher of Director of Jinsoo Chung, Prostate Cancer Center, National Cancer Center, Goyang, Korea. He has published more than 30 papers in reputed journals.
Abstract:
Multilocular cystic renal neoplasm of low malignant potential (MCRNLMP) and multicystic renal cell carcinoma (MCRCC) are morphologically indistinguishable. MCRNLMP is a tumor composed entirely of numerous cysts, the septa of which contain individual or groups of clear cells without expansive growth. However, unlike MCRCC, neither recurrence nor metastasis have been reported in MCRNLMP. The aim of this study was to identify significant differential pathological characteristics in resected specimens from patients with MCRNLMP (n = 13) and MCRCC (n = 17) using immunohistochemistry of 25 tissue markers. Staining interpretation was performed semi-quantitatively using the H-score (0–300) or intensity score (0–3), and differences between groups were evaluated using the Fisher exact and Wilcoxon rank-sum tests. During a median follow-up of 66.2 months (1–141.9 months), there was only one case of recurrence in MCRCC among 30 patients. There were 19 (63.3%) at stage pT1a, 8 (26.7%) at stage pT1b, and 3 (10%) patients at stage pT2. Tumor necrosis rate (0% vs. 52.9%) and median tumor size (3.2 cm vs. 4.1 cm) significantly differed between MCRNLMP and MCRCC samples. Among the 25 tissue markers, only HIF1a, PDGFRα, SMA, VEGFR1, VEGFR2, VEGFR3, CD10, CD31, CD34, CK7-tubule, TGAse-2, and Ki-67 showed significantly different expression between the groups. These tissue markers with differential expression between MCRNLMP and MCRCC can provide a clue to understanding their distinct pathophysiology.
Biography:
Juliana Guimarães Fonseca is a Biotechnologist graduated from Federal University of São Carlos, Brazil, in 2012. She has Master’s degree in Science from University of Sao Paulo, in 2014. Currently, she is PhD candidate at University of Sao Paulo and her research is on proteomics, more specifically, characterization of the protein profile of contaminating bacteria present in the first-generation ethanol fermentation process by MALDI TOF. She has already published papers in reputed journals and wrote book chapters.
Abstract:
Bioethanol has gained space in the global energy sector in recent years, wherein Brazil is the second largest producer of this fuel. The process responsible for ethanol production is fermentation, in which sugars available in sugarcane are turned into alcohol by fermenting microorganisms. Nonetheless, large-scale fermentation does not occur in an aseptic environment. In this way, presence of contaminants, mainly acid-lactic bacteria (LAB), is a recurring problem in the process, which can cause a decrease in ethanol yield. Thus, identification of contaminating bacteria by mass spectrometry techniques allows a rapid and efficient identification of contaminating microorganisms, which can prevent drastic falls or interruption of fermentation process. We isolated and identified contaminating microorganisms from fermentative process by Sanger sequencing of gene 16S rRNA. We also characterize protein profile of these bacteria through matrix-assisted laser desorption/ionization (MALDI). We identified 13 bacteria that belonged to Lactobacillus genus. We also optimized the methodology used for MALDI to identify LAB from a small number of bacteria grown in MRS media using a 1 μL loop and suspended in 10% formic acid. For the identification of protein characteristic spectral profile, the best results were obtained when 1 μL of the mixture was spotted onto a polished steel target plate and overlaid with 0.5 μL of ethanol. The matrix used was saturated solution of α-cyano-4-hydroxycinnamic acid 50% acetonitrile – 2.5% trifluoroacetic acid. This is the first time that mass spectrometry was used to identify bacterial contaminants from fermentation tanks in large scale ethanol production.
Biography:
Siti Sarah Hamzah is a final year PhD student at University of Warwick, United Kingdom. She received a Bachelor degree in Biochemistry and Molecular Biology from The University of Melbourne, Australia and an Honours degree in Pharmaceutical Sciences from Monash Institute of Pharmaceutical Sciences, Australia. After 2 years as Medical Scientist in the Biochemistry Dept. of Institute for Medical Research, Ministry of Health Malaysia, she is now serving in the Endocrinology Dept. of the same institute. Her current research involves study of the role of alternative splicing (AS) in epithelial-to-mesenchymal transition (EMT) in breast cancer models.
Abstract:
Studies show that 70% of breast cancers are estrogen receptor-positive (ER+) and activation of cellular processes involved in several tumorigenesis-associated events are detected predominantly in ER+ breast cancer. To characterize the global proteomic alterations affected by sex hormone estrogen, a label-free quantitative proteomic method was used to identify and quantify the differentially expressed proteins in the hormone-responsive MCF7 cells exposed to 17b-estradiol (E2) for 24 h. Using a selection filter cut-off of >2-fold change and p value<0.01, 3,000 proteins were identified: 93 of them were upregulated whereas 64 were downregulated by E2. The list of upregulated proteins included the 91 kDa serine-arginine protein kinase 1 (SRPK1), a key kinase involved in the phosphorylation of serine-arginine proteins (SRps) during pre-mRNA alternative splicing that also found to be overexpressed in malignant cancer cells. Follow up studies employing qRT-PCR and Western blots revealed increased phosphorylation signal of SRp40 and SRp30 as well as increased production of aberrant CD44 mRNA isoforms. In cellular models of ER+ breast cancer, SRPK1 expression was also positively regulated by corticotropin-releasing hormone (CRH) a hypothalamic hormone involved in adaption to stress. In contrast, proteome analysis using Nano-flow Ultra HPLC coupled to Orbitrap fusion demonstrated that CRH did not affect SRPK1 expression in ER-breast cancer and phosphosites analysis in cells depleted with SRPK1 kinase resulted in reduced detection of signal specific for phosphorylation site of SRp40 and SRp30. Altogether, these results identify potential targets of hormone-altered proteome profiles in different types of cancer cells hence triggering signaling networks that could enrich proteome diversity by producing distinct oncogenic protein isoforms.
Kyoungsook Park
Sungkyunkwan University, South Korea
Title: Analysis of differential expression of glycosyltransferase in mouse liver
Biography:
Kyoungsook Park has completed her PhD at the University of Pennsylvania, Philadelphia, USA.She is a Research Professor at Sungkyunkwan University in Republic of Korea and she has published many papers in reputed journals and has been serving as an active Member of AACR and a representative Committee Member of KSBMB.
Abstract:
Glycosylation is a major post-translational gene regulation and is involved in many aspects of cellular processes. It plays pivotal functional roles in immune surveillance, inflammation, and drug responses among many other biological processes. Establishment of a mouse model which mimics a human glycosylation profiling is urgent and is instrumental to accelerate the non-clinical tests prior to the human clinical trials for a potential novel drug candidate. To this end, we set out to identify the glycosylation-regulating genes (GRGs) which were differentially regulated in human liver compared to the mouse counterpart. Among major organs, we chose a liver due to its critical roles in metabolism. To facilitate the screening of differentially expressed GRGs, Glycosylation Profiler PCR array expression panel was utilized as an initial screening approach with the total RNAs derived from commercially available normal human liver tissues and B6 mouse liver tissues. Candidate GRGs were further confirmed by RT-PCR analysis with individually designed specific primer sets. Our findings would provide crucial information for construction of a non-clinical mouse model with humanized glycosylation profiling, which is critical for cell-cell interaction and signaling in immune responses, and drug responses.
Sulhee Kim
Korea University, South Korea
Title: Structural analysis of tegumental allergen-like proteins from Schistosoma mansoni
Biography:
Sulhee Kim is a PhD student at Biosystems & Biotechnology Department of Korea University in South Korea. Recently, she have published more than 5 papers in reputed journals, such as Autophagy, Nucleic acids research, and so on.
Abstract:
Schistosomiasis (or bilharzia) is the most common cause of death from a parasitic disease after malaria. The disease can be treated effectively with the drug praziquantel (PZQ). The tegumental allergen-like (TAL) proteins from Schistosoma mansoni (SmTAL) are part of a family of calcium binding proteins found only in parasitic flatworms. These proteins have attracted interest as potential drug or vaccine targets, yet comparatively little is known about their biochemistry. For elucidating the exact mechanism of drug-interaction with tegument associated proteins from parasites (TALs), here, we had grown the suitable crystals of TALs from Schistosoma mansoni. The crystals were diffracted at 2.1 Å and 2.5 Å at synchrotron radiation, respectively. The spacegroup of smTAL1 and smTAL2 are hexagonal system (P64 and P6422), respectively. They consist of two domains, calmodulin-like (EF hand) and dynein-like (DLC domain). We will discuss the difference between smTAL1 and smTAL2.
Recent Publications
-
P Steinmann, J Keiser, R Bos, M Tanner and J Utzinger (2006) Schistosomiasis and water resources development: systematic review, meta-analysis, and estimates of people at risk. Lancet Infect. Dis. 6(7):411e425.
- J Utzinger, S L Becker, S Knopp, J Blum, A L Neumayr, J Keiser, C F Hatz (2012) Neglected tropical diseases: diagnosis, clinical management, treatment and control. Swiss Med. Wkly. 142:w13727.
- D Cioli, L Pica-Mattoccia, A Basso and A Guidi (2014) Schistosomiasis control: praziquantel forever? Mol. Biochem. Parasitol. 195:23e29.
Radostina Velikova
Bulgarian Academy of Sciences, Bulgaria
Title: Proteomics analysis of antitumoractivity of Helix and Rapana hemocyanins
Biography:
Ms. Radostina Velikova is currently she is working in the Institute for Organic Chemistry with Center for Phytochemistry, Bulgarian Academy of Sciences.
Abstract:
Hemocyanins (Hcs) are copper-containing glycoproteins that act as oxygen transporting proteins in many arthropods and mollusk species. Hemocyanins from the molluscs Helix aspersa (HaH), Helix lucorum (HlH) and Rapana venosa (RvH) exhibiting different oligosaccharide structures have been investigated for potential use in therapy of bladder cancer permanent cells,. In vitro studies on the antitumor activities of these proteins were performed in T-24 cells and compared to doxorubicin and mitomycin-C. Control experiments were performed using normal urothelial HL 10/29 cells.
The obtained results show that the human tumor T24 cell lines are sensitive to the action of the tested hemocyanins and their isoforms. The inhibition of the tumor cell growth was dose and time dependent and was observed after incubation with native HaH and HlH, and FUs (βc-HlH-h and RvH-c. Cells treated with both FUs, βc-HlH-h and RvH-e, showed apoptotic and necrotic cells and this inhibition was stronger than the effect measured for doxorubicin treated cells. No growth inhibition of the normal urothelial cell line HL 10/29 was observed after treatment with HlH, HaH, RvH and their isoforms. The impact of hemocyanins on tumor cells was investigated by 2D-gel PAGE and several proteins showed indeed altered abundancies. The most effective inhibition of tumor cells is probably caused by a specific novel and unusual N-glycan oligosaccharide structure on HlH with methylated hexoses, an internal fucose residue connecting one GalNAc (ß1-2) and one hexuronic acid.
Ondrej Pastva
Institute of Hematology and Blood Transfusion, Czech Republic
Title: The original SPR-based Hsp70 biosensor for rapid quantification and revelation of Hsp70 substrates in complex solution
Biography:
Ondřej Pastva is a PhD student in the Biochemistry group at the Institute of Hematology and Blood Transfusion, Prague, Czech Republic. His main research interest is in utilizing of optical biosensors and bioassays for improving of diagnostics in Leukemias.
Abstract:
The Hsp70s (heat shock 70kDa) proteins are ubiquitous molecular chaperones forming the center of protein folding, refolding, and trafficking in all organisms. Hsp70 interacts with hydrophobic peptide segments of non-folded chains, as well as near-native, misfolded, and aggregated proteins (clients), both at normal and at stressed conditions. To date, mostly the ADP-affinity chromatography or affinity purification chemical tool is generally used to detect the Hsp70 substrates. These techniques are time consuming and samples may contain non-specific ADP-like proteins or should be modificated before subsequent identification through biochemical techniques. We designed a new assay which allows real-time monitoring of the (i) trapping and retention of Hsp70 client proteins from blood plasma samples and (ii) highly efficient elution (>90%) of retained proteins. The Hsp70 trap assay has been implemented using a multichannel surface plasmon resonance (SPR) biosensor functionalized by Hsp70. Trapping and elution of client proteins is controlled by ATP/ADP exchange. The nucleotide and cations requirements were investigated and set up. In the three-step assay, the reference channel represents non-specific proteins was designed to control of elution. The SPR biosensor was used for the screening of healthy controls and clinical samples – myelodysplastic syndromes patients (MDS, subgroups – RARS, RAEB, and AML). We observed significant differences between the tested groups and that the amount of misfolded proteins correlated with the progression of the disease. Our results are in a good agreement with the fact that oxidative stress as one of the main factor in MDS disease progression leads up to covalent modifications that destabilize and inactivate proteins. To the best of our knowledge, this is the first biosensor using Hsp70 to reveal a misfolded protein complexes. The Hsp70 trap assay seems to be a promising tool for general profiling of disease pathogenesis and progression. Using three-step assay, Hsp70 client proteins were detected in blood plasma in ~35 min at 25 ng/cm2 to 150 ng/cm2 concentration for healthy controls and AML patient, respectively.
Abdullah Essa Alsubhi
King Abdulaziz University, Saudi Arabia
Title: Proteomics analysis of proteins associated with urinary tract infections
Biography:
Abdullah Essa Alsubhi completed hisBachelor’s Degree from Umm Alqura, KSA in 2011 and Master’s Degree from King Abdulaziz University, Faculty of Applied Medical Sciences, Department of Clinical Laboratory Science. He have been working in Saudi Health Ministry for six years. He has worked his practical master research in Aberdeen University, Scotland.
Abstract:
Urinary tract infection (UTI) is one of the most prevalent bacterial infections in the world. It affects urinary tract system including bladder and kidney. Gram-negative bacteria is a major cause, particularly Escherichia coli (E. coli), which is considered a main causative agent for 80-90% of community-acquired infection and for about 40% of nosocomial UTI. Moreover, it is responsible for 25% of recurrent infection. The study of the genome identified and sequenced three E. coli strains causing UTIs and all strains showed higher expression of certain proteins; such as adhesion protein p, and toxin such as hemolysin and high expression of proteins responsible for iron acquisition. Proteomics is used to analyze and identify complete components of proteins and it can be used to distinguish between bacteria based on synthesized protein. In addition, proteomic is applicable to identify possible target of therapy. This study aims to compare protein profiles of E. coli from different UTI patients and identify possible unique proteins signature for future biomarker studies. Seven urine samples were taken from different UTI patients and the pathogenic agent among those patients was E. coli. Urine E. coli isolates were analyzed by 1D SDS-PAGE, 2DGE and LC-MS/MS and three interesting spots were selected for protein identification by LC-MS/MS. Many significant differences were observed in protein profiles of E. coli isolates in both 1D SDS-PAGE and 2DGE regardless of type of E. coli species. Finally, it was concluded that E. coli isolates obtained from the different UTI patients had different protein profiles.
Joanna Tracz
Poznan University of Medical Sciences, Poland
Title: Analysis of the leukocytes proteome in atherosclerosis related and non-related to chronic kidney
Biography:
Joanna Tracz has received his/her Bachelor’s degree in 2015 and completed his/her Master’s degree in Chemical Science in 2016 and Biological Science in 2017. Presently, She/he is a PhD student in Laboratory of Mass Spectrometry at the Polish Academy of Sciences. She/he is involved in project concerning the molecular mechanism of atherosclerosis progression in chronic kidney disease.
Abstract:
The major cause of mortality in patients with chronic kidney disease (CKD) is atherosclerosis related to traditional and non-traditional risk factors. However, the understanding of the molecular specificity that distinguishes the risk factors for classical cardiovascular disease (CVD) and CKD-related atherosclerosis (CKD-A) is far from complete. Although dyslipidemia is common in CKD patients, epidemiological data show that in CKD the link between cholesterol and its fractions is not as straightforward like in the general population. CKD is frequently accompanied by reduced of plasma HDL concentrations, and normal or even low serum total cholesterol and LDL concentrations. Normal HDL function is reverse cholesterol transport from peripheral cells and its transport to the liver. HDL protects of LDL against oxidation and suppress of systemic inflammation. Therefore, HDL deficiency is the key in perpetuating chronic inflammation and oxidative stress leading to atherosclerosis. On the other hand, it is suggested that lipid abnormalities in CKD are characterized by more qualitative abnormalities and may be related to HDL function rather than HDL deficiency. Mass spectrometry-based proteomic analysis is excellent, powerful tool for tracking molecular changes during progression of CKD. In this study we investigated the alterations in leukocytes protein accumulation in patients with CKD and classical cardiovascular disease (CVD) without CKD. Cells collected from patients in various stages of CKD, CVD patients without symptoms of kidney dysfunction and healthy volunteers (HVs), were analyzed by a label-free proteomic approach. Obtained cells were also analyzed in term of inflammation modulators and the oxidative status enzymes. Label-free quantitation analysis revealed characteristic proteins of particular stage of atherosclerotic plaque formation process and CKD progression. All proteomic data were subjected to bioinformatic analysis for identification of specific for CKD and CVD pathways. Further research should focus on precise profiling of metabolites and/or lipids present in cells during the atherosclerosis development.
- Mass Spectrometry in Proteome Research | Proteomics in Biochemistry and Molecular Biology | Proteomics and Molecular Medicine
Location: Paris, France
Session Introduction
Lungu N Claudiu
Babes-Bolyai University, Romania
Title: MSCM-Multiple selective proteo nano cluster mesh coating CD’s agonist/antagonist
Time : 10:00-10:20

Biography:
Lungu N. Claudiu is a last year PhD in organic chemistry at “Babes-Bolyai” University, Cluj, Romania. He has a degree in medicine (MD) and in pharmacy. His areas of interest are: computational chemistry, drug discovery and materials science. He wrote a number of papers regarding nano bio assemblies, polymers, bio nano interactions and quantitative structure activity relationship (QSAR). In the last time, his areas of interest are bioinformatics and bioactivity study. He delivered several oral presentations in: Berlin (Germany), Warsaw (Poland) and Bergen (Norway) regarding nano assemblies, as a part of an European research grant in the fiel of nanoscience.
Abstract:
Cell surface molecules (CSM) like L selectin, VLA-4, LFA-1, CD2, CD4, TCR, CD44, CD45RA and CD45RO play a major role in cell interaction, signalling and function. CSM are in a “mosaic” of activated–inactivated states. These states determine cell behaviour. Cells are controlled by a “cocktail” of molecules that act on CSM modulating their states. Nano system was designed using computational methods like molecular dynamics, docking, ligand design, virtual screening and experimental methods like isothermal titration calorimetry (ITC) and single cell imaging techniques. The system is composed of nano polymers, graphene and fullerene patches on witch wild or synthetic “ligands”-amino acids or small peptide for CSM are incorporated. Physico chemical triggers were used to activate MSMC. Cellular meshing by MSCM allows controlling and guiding of cellular dynamic interaction with all the pathways involving the cell. MSMC by its nano polymer mesh can protect or stabilize a certain cell or tissue. MSMC was made cell specific by incorporating Ig motifs against different types of cells. Computational models of L-selectin, VLA-4, LFA-1, CD2, CD4, TCR, CD44, CD45RA and CD45RO were designed and incorporated into a nano polymer mesh. MSMC outer surface was coated with phosphatidilinositol, sphingnomielin and cholesterol mono layer. The system goal is to be administrated iv. In the first stage of development, the attention was focused on single cell manipulation. The viability of the cell (unintended cell death) and the lack of adverse effects like aggregation, hemolysis4, toxicity were tested. Each receptor was characterized individualy and a series of proteic motifs were developed for incorporation into MSMC. In conclusion, a bio-nano-device5 was designed to control a cell by stimulating/inhibiting the CD’s. System was designed to be cell specific and tissue specific; device is ,,controlled’’ by physico-chemical stimuli. The ultimate goal of this system is to control large volumes of cells eventually tissues.
Aviwe Ntsethe
University of KwaZulu Natal, South Africa
Title: Isolation and quantification of nanovesicles in isoproterenol-induced myocardial infarcted rats
Time : 10:20-10:40

Biography:
Aviwe Ntsethe has completed his Honour’s degree in Medical Science (Physiology) from University of KwaZulu Natal, School of Laboratory Medicine and Medical Science. He is now doing his Master’s degree in Physiology in the same university.
Abstract:
Myocardial infarction (MI) is one of the leading causes of death worldwide. The pathogenesis and aetiology of MI is still unclear. Cardiac troponin is the only known cardiac-specific marker for the diagnosis of MI but due to the delayed release of troponin in the circulation, a novel cardiac biomarker is needed in the early stages of development of MI to reduce MI mortality. Exosomes are reported to be highly regulated by stress. Thus we assessed the hypothesis that exosome secretion is increased following MI and thereby serve as biomarker for MI. The aim of this study was to quantify exosomes in an isoproterenol (ISO)-induced MI rats. Twelve rats were used in this study, Group-A (n=6) was the normal control rats and group-B (n=6) was the ISO-treated group. Group-B animals were injected with isoproterenol for two consecutive days to induce MI. Blood pressure, heart rate and body weight were monitored in all animals prior the ISO injection and throughout the experiment. After the second ISO-injection, all animals were sacrificed and blood, heart tissues were obtained. Histopathological analysis was performed in heart tissue samples and levels of cardiac markers were measured from the serum. Exosomes were isolated from the plasma and quantified by differential ultracentrifugation, nanoparticle tracking analysis, transmission electron microscope and ELISA respectively. MI was confirmed by the increase in BP and cardiac markers in ISO-induced rats. The concentration of exosomes was elevated in plasma of ISO-treated animals. The study revealed that exosomes are potential biomarkers of myocardial infarction.
Laura Ion
Alexandru Ioan Cuza University, Romania
Title: Proteomic-based approaches for the characterization of peptides associated with Alzhemier’s disease
Time : 11:00-11:20

Biography:
Laura Ion has completed her PhD at Alexandru Ioan Cuza University of Iasi, RO and currently she is a Postdoctoral Fellow at the same university. Her research area is focused on peptide and protein chemistry by using SDS-PAGE electrophoresis, mass spectrometry, liquid chromatography, etc. During her studies, she obtained several research stages at Konstanz University and Steinbeis Center of Biopolymer Analysis and Biomedical Mass Spectrometry in Rüsselsheim, Germany.
Abstract:
Currently, the development of proteomic approaches (e.g. mass spectrometry, electrophoresis) became a key tool for characterization of non-covalent bio-molecular complexes, such as those with heavy metal ions, small organic molecules or even in presence of sodium dodecyl sulfate (SDS). Moreover, various proteins can form oligomers (ex. Amyloid beta peptide) and the relationship between different states of proteins (starting with monomeric to oligomeric state) is important for the protein activity. Therefore, a critical step for a better understanding of the cell is to determine the protein complexes structure. Therefore, our research work was focused on conformational studies by using small peptides, such as tetraglycine, newly model peptides (histidine-containing Ala- and Gly-based peptides) and large peptides, such as amyloid beta peptide under various environmental conditions (ex. SDS, stearic acid, metal ions, ammonium acetate or trifluoroethanol solution). Our results show that (i) SDS is able to reduce the proportion of peptide oligomers; (ii) SDS solution added to the Aβ solution severely changes the peptide conformation, with disappearance of the β-sheet conformers and doubling the proportion of β-turn isomers; (iii) on investigating histidine containing peptides, the proportion of conformers was found dependent on the amino acid sequence.
Leila Nasehi
Zanjan University of Medical Sciences, Iran
Title: Stable silencing of IGF1R using lentiviral-mediated shRNA and evaluating sensitivity to immunotherapy with monoclonal antibody rituximab in lymphoma cells
Time : 11:20-11:40

Biography:
Leila Nasehi received her Master’s in Microbiology in 2008 and PhD degree in Molecular Medicine in 2017 from Tehran University of Medical Sciences. Her research activities take place in the field of antibiotic resistance in patient’s samples, particularly in the field of resistance genes in microorganisms (ESBL), and genetic manipulation by employing plasmids or lentiviruses vectors for silencing or stable silencing in adherent & suspension cells. She is a faculty member of Zanjan University of Medical Sciences.
Abstract:
The high expression of insulin-like growth factors and their receptors is a common phenomenon in the process of invasion and metastasis of many malignancies such as lymphoma. Rituximab is the first monoclonal antibody to be approved for the treatment of lymphoma. Epitope complex of rituximab has been determined by co-crystallizing a synthetic peptide mimic of the extracellular loop epitope of CD20 (residues 163–187) in complex with the antigen-binding fragment of rituximab. Genetic mutations in the rituximab epitope can reduce the binding and efficacy of the antibody and these mutations are important causes of failure to treatment. Sequencing and bioinformatics analysis exhibits mutations in CD20 gene indicative of resistance and therefore it seems the synergistic effect of combined therapy with the stable system of lentiviral gene therapy and immunotherapy can provide a promising therapeutic approach for the treatment of lymphoma. We made lentiviruses using six lentiviral cassettes against IGF-IR and used to transduct HEK293T cells. Then we compared them to non-silencing control cassette and the reduction in mRNA and protein expression level was determined using real time PCR and Western Blot techniques. The most effective cassette was selected and used to transduct Raji cells. The expression of IGF-IR and Bcl2 at mRNA and protein level was assessed and the cell growth and cytotoxicity were assessed using MTT assay. We evaluated the role of IGF-1 receptor in growth and proliferation of Raji tumor cells by reducing the expression of IGF-IR. Finally, we detected sensitivity to rituximab- immunotherapy in the lymphoma cells.
Susan Sabbagh
Dezful University of Medical Sciences, Iran
Title: A proteomic analysis on human sperm tail: comparison between normozoospermia and asthenozoospermia
Time : 11:40-12:00
Biography:
Susan Sabbagh is an educated physiotherapy at Shiraz University of Medical Science and she has post graduated from anatomical science in Ahwaz University of Medical Science. Since then she started teaching at Dezful University of Medical Science as a faculty member. Her main research was on neurology field and done some researches on multiple sclerosis, neural tube defects and hearing loss and published 8 papers in reputed journals. From 2014, she was attracted to proteomics and learned about it. Her project on sperm tail proteomics was published in journal of assisted reproduction and genetics on 2015.
Abstract:
Introduction: Asthenozoospermia is a common cause of human male infertility characterized by reduced sperm motility. The molecular mechanism that impairs sperm motility is not fully understood. This study purposed to identify novel biomarkers by focusing on sperm tail proteomic analysis of asthenozoospermic patients.
Materials & Method: Sperm were isolated from normozoospermic and asthenozoospermic semen samples. Tail fractions were obtained by sonication followed by Percoll gradient. The proteins were extracted by solubilization and subjected to two-dimensional gel electrophoresis (2-DE); then, the spots were analyzed using ImageMaster 2D Platinum software. The significantly increased/decreased amounts of proteins in the two groups were exploited by matrix-assisted laser desorption-ionization time-of-flight/time-of-flight (MALDI-TOF-TOF) mass spectrometry.
Results: Three hundred and ninety protein spots were detected in both groups. Twenty-one protein spots that had significantly altered amounts (p<0.05) were excised and exploited using MALDI-TOF-TOF mass spectrometry. They led to the identification of the following 14 unique proteins: Tubulin beta 2B; glutathione S-transferase Mu 3; keratin, type II cytoskeletal 1; outer dense fiber protein 2; voltage-dependent anion-selective channel protein 2; A-kinase anchor protein 4; cytochrome c oxidase subunit 6B; sperm protein associated with the nucleus on the X chromosome B; phospholipid hydroperoxide glutathione peroxidase-mitochondrial; isoaspartyl peptidase/L-asparaginase; heat shock-related 70 kDa protein 2; stress-70 protein, mitochondrial; glyceraldehyde-3-phosphate dehydrogenase, testis-specific and clusterin.
Conclusion: Fourteen proteins present in different amounts in asthenozoospermic sperm tail samples were identified, 4 of which are reported here for the first time. These proteins might be used as markers of male infertility, targets for male contraceptive development, and to predict embryo quality.