Day 1 :
Keynote Forum
Boris Y Zaslavsky
Cleveland Diagnostics, USA
Keynote: Properties of membrane-less organelles and aqueous two-phase systems
Time : 9:35-10:05

Biography:
Boris Y Zaslavsky is a Bioanalytical Chemist. He graduated from Moscow State University in 1967, PhD in 1972; DSc in 1985 from USSR Academy of Sciences, Moscow, USSR. From 2012-present, he is a Chief Scientific Officer of Cleveland Diagnostics, Cleveland, OH, from 1997-present, he is a Vice President, Director of Research and Cofounder of Analiza, Inc., Cleveland, OH. He has over 180 publications in peer-reviewed journals, 1 monograph, over 10 USA and international patents issued. His research interests are: Clinical proteomics, role of water in biology, and aqueous two-phase systems.
Abstract:
Coordination of numerous cellular biochemical reactions in space and time is achieved by compartmentalization. In addition to intracellular membranes acting as physical barriers for several cellular organelles there is a multitude of membrane-less organelles formed by liquid-liquid phase separation. The principles governing phase separation and functions of such organelles in vivo are poorly understood as of now. However, the much better studied aqueous two-phase systems formed by two polymers may serve as a model of membrane-less organelles. Such systems originate from polymer influence on the solvent properties of water. The phase forming polymers may include proteins and polysaccharides. The differences between solvent features of aqueous media in the two phases may be quantified and manipulated by polymers’ concentrations and additives of inorganic salts or small organic compounds, such as sucrose, sorbitol, etc. The differences between electrostatic properties of the phases as well as those between solvent features may be quantified using partitioning of homologous series of charged compounds and solvatochromic dyes as molecular probes for the solvent dipolarity/polarizability, solvent H-bond donor acidity, and solvent H-bond acceptor basicity. The differences between solvent features and electrostatic properties of the phases govern unequal distribution of proteins and other natural compounds in aqueous two phase systems and in membrane-less organelles. This solvent-driven partitioning, and not the “normal” protein-protein interactions, might cause enrichment of some proteins within the membrane-less organelles. It will be shown that proteins may influence solvent features of water and their effects are similar or exceeding those displayed by common macromolecular crowding agents and organic osmolytes. It is suggested that the effects of proteins on the solvent features of aqueous media may regulate the phase separation in vivo.
Keynote Forum
Bin Huang
Kaohsiung Medical University, Taiwan
Keynote: Integrative analysis of post-translational protein S-nitrosylation in endothelial cells
Time : 10:05-10:35

Biography:
Bin Huang obtained his PhD degree from the Department of Plant Science, National Taiwan University. He then focused on Cardiology during his Postdoctoral studies. He became experienced in the gaseous molecule-mediated post-translational proteome, particularly in the NO-mediated S-nitrosylation when studying endothelial cells of the vascular system. He became interested in the behavior of mitochondrial fusion/fission when studying cell aging and drug-resistance of cancer cells. In addition to these research interests, he develops the Center for Stem Cell Research of Kaohsiung Medical University as the Vice Director.
Abstract:
Nitric oxide (NO), an endogenous evolutionary gaseous molecule with labile character can bind to cysteine residues (Cys-NO, S-nitrosylation) and then alter the enzyme activity. NO is regarded as a mild reactive oxygen/nitrogen species (ROS/RNS) that can compete with other, more potent ROS/RNS, and protects cells from irreversible oxidative stress caused by free radicals. At present available methodologies applied to study the implications of NO in physiological responses include Western blotting to measure the phosphorylation of endothelial nitric oxide synthase (eNOS) at Ser1177 and Ser633 residues and detecting gaseous NO by Griess reagent. However, this reagent is greatly affected by the presence of peroxinitrite (ONOO−). Therefore, the new fluorescent probe - 5-amino-2-(6-hydroxy-3-oxo-3H-xanthen-9-yl) benzoic acid methylester (FA-OMe) - that specifically binds to endogenous NO, was developed and utilized. As a result, the elevated production of NO can be estimated not only by eNOS phosphorylation in Western blots but also by direct quantification using FA-OMe. Once it became possible to confirm the production of NO, the identification of the subsequent protein S-nitrosylation resulted from NO binding to the cysteine residues became important. The utilizations of commercialized antibody and mass spectrometric devices were reported to detect the Cys-NO residue directly. However, for the reason of poor antibody specificity and weak chemical binding of Cys-NO, both two methods were not reliable. We therefore designed a tag-based labeling system on cysteine residue that modified from biotin-switch, (e.g. IAA, IAM and iTRAQ). Cys-NO will be replaced by these tags and was then detected either by 2-DE-based Western blot or mass spectrometry with identical molecular weight shifts. The whole profiles of enzyme activation, gaseous NO molecule production, and the subsequent protein S-nitrosylation could be analyzed simultaneously to explain more details about the physiological mechanisms of action in protein S-nitrosylation.
Keynote Forum
Manuel Gea
Bio-Modeling Systems, France
Keynote: The future of systems medicine: Everything you always wanted to know about the “reality†of big data and AI “big mirages†but were afraid to ask
Time : 10:55-11:25

Biography:
Manuel Gea is a Serial Entrepreneur, Co-Founder of the world’s first Mechanisms-Based Medicine Company, thinking & doing out of the box applying “general semantics” principles and Augmented Intelligence vs. Artificial Intelligence. He is a Keynote Speaker, independent board member, expert & business angel developing disruptive innovations (technologies, novel therapies & business models) in the life sciences, IT, pharma, healthcare & cosmetic sectors. He is graduated from Ecole Centrale Paris in operational research and data integration, and has Sociology of Organizations degree from Paris IX Dauphine University. He is also Chairman of the Independent trans-discipline biotech Think Tank Adebiotech, and the Co-founder and Chairman of Centrale-Santé, the French Health Think-Tank gathering 2500 members, professional involved in sector innovations and creating value for patients. He is Co-founder, CEO & VP R&D IT of BMSystems, dedicated to the discovery of cost-effective new therapeutic, diagnostic & preventive solutions.
Abstract:
Statement of the Problem: With a 95% failure rate, the Big Pharma R&D model focused on testing new patentable compounds for novel targets based on KOL concepts is not more pertinent for at least 3 reasons: 1) Despite decades of investments in Omics technologies and Systems Biology programs produced few relevant results due to 3 “side effects” and a conceptual mistake: Life mechanisms are complex not complicated! 2) The “mirage” of Artificial Intelligence (AI) that MUST follow rules in a world where humans massively do not. 3) The unreliability of scientific and clinical publications is increasing, and the valuable negative results are not published. Why the transfer of the digital tools and methods does from the internet world to the life sciences R&D world does not work properly. One reason is that the founding basements of the two worlds do not obey the same rules. The challenge is not a question of technologies only, its needs to redefine discovery concepts.
The Future of Systems Medicine: 1) Understanding and validating the mechanisms of a disease/disorder becomes the first objective. 2) Finding the most adapted solutions is a necessary consequence. The Mechanisms-Based Medicine Concept gives the basements to apply CADI™ (Computer Augmented Deductive Intelligence) Discovery that addresses life mechanisms complexity and the unreliability of scientific and clinical publications by combining the strengths of human and artificial intelligences in the right order.
The 5 CADI™ Discovery Principles: 1) Mechanisms-Based Medicine Concept; 2) Architectural Principle; 3) Negative Selection Principle; 4) Steps Validation Principle; 5) Integrated Solutions Principles.
Conclusions: CADI Discovery already led to a world’s first in neurodegenerative diseases, 1 therapeutic spin-off and 1 exclusive our license, 4 issued patents, 10 publications, and 39 CADI™ programs among completed or open for collaboration.

Recent Publications:
1. F Iris F, D Filiou, Ch W Turck (2014) Differential proteomics analyses reveal anxiety-associated molecular and cellular mechanisms in cingulate cortex synapses. American Journal of Psychiatry and Neuroscience 2(3): 25-42.
2. Iris F (2012) Psychiatric systems medicine: closer at hand than anticipated but not with the expected portrait. Pharmacopsychiatry 45 (Suppl. 1): S12–S2.
3. Turck CW, Iris F (2011) Proteome-based pathway modelling of psychiatric disorders. Pharmacopsychiatry 44 (Suppl 1): S54-61.
4. Iris F, Gea M, Lampe PH and Santamaria P (2009) Production and implementation of predictive biological models. Med Sci. 25: 608-16.
5. Iris F, Gea M, Lampe PH and Santamaria P (2009) Production and implementation of predictive biological models. Med Sci. 25: 608-16.
- Mass Spectrometry in Proteome Research | Protein Expression and Analysis | Proteomics from Discovery to Function | Molecular Medicine and Diagnostics
Location: Paris, France
Session Introduction
Oliva Petra
Sanofi Genzyme, USA
Title: Strategy for determining clinical biomarker panel
Time : 11:25-11:45

Biography:
Petra Oliva has 10 years of biotech experience and close to 20 years scientific expertise in an advanced interdisciplinary research in analytical chemistry, biotechnology and biochemistry with deep understanding of biomarker discovery, qualification and validation. She is currently Pr. Scientist at Translational Science group within Sanofi, US. She is managing analytical/bioanalytical support from pre-clinical stage, through PhI & PhII all the way to the commercial phase. Petra and her team established several innovation platforms (as targeted proteomics & MS imaging) winning multiple global Sanofi awards. She also has 8 years collaboration with CDC on LSD newborn screening.
Abstract:
Biomarkers are essential for improving the outcomes of clinical trials and accelerating drug development. Mass spectrometry (MS)-based proteomics applied early to clinical samples has the potential to identify and narrow down predictive and pharmacodynamics biomarkers. These markers can then be used in clinical trials for patient stratification and to increase sensitivity of primary endpoints for a better measurement of therapeutic response. Unbiased proteomic profiling is powerful during the exploratory biomarker stage for monitoring hundreds or thousands of proteins, but throughput is low and relative quantitation is variable for low abundant analytes. Here, we describe an integrated, hypothesis-driven strategy that combines unbiased proteomics and literature mining to generate a highly quantitative and reproducible targeted proteomics assay for testing in large, representative patient cohorts for candidate biomarker screening. Combined with appropriate statistical and bioinformatics processes, this strategy will facilitate selection of a robust biomarker panel which may be validated as a companion diagnostic or as a clinical tool.
Ida Chiara Guerrera
INSERM, France
Title: Proteomics of rare genetic diseases: Impact of cystinosin mutations on protein stability and protein network
Time : 11:45-12:05

Biography:
Ida Chiara Guerrera (PhD, UCL London) is Head of the Proteomics Laboratory at the Necker Hospital in Paris. Her interests center on understanding human disease by applying innovative proteomics approaches to biomedical research. She participates in fundamental research to understand mechanisms underlying disease and translational research for biomarker discovery. Her work has contributed to exploring three rare genetic diseases: cystic fibrosis, cystinurie and cystinosis.
Abstract:
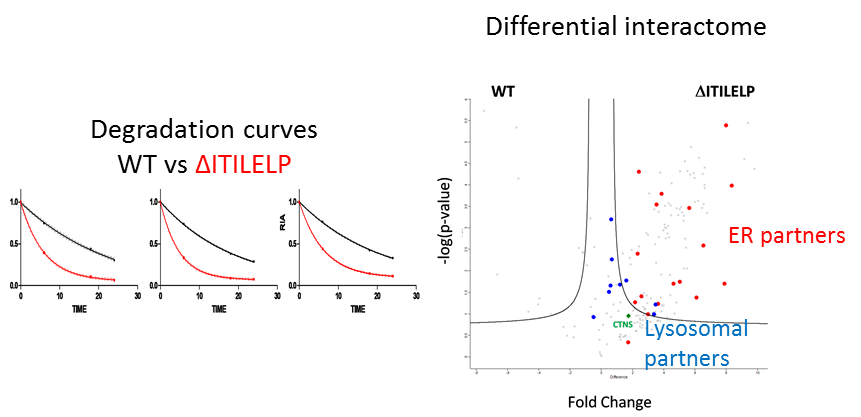
Statement of the Problem: Cystinosis is a rare autosomal recessive storage disorder characterized by defective lysosomal efflux of cystine due to mutations in the CTNS gene encoding the lysosomal cystine transporter, cystinosin. Over 100 mutations have been reported, leading to varying disease severity, often in correlation with residual cystinosin activity as a transporter. However, the correlation between genotype and phenotype is not always clear and we applied different proteomics approaches to better understand the mechanisms of this disease. To unveil additional roles of cystinosin, we studied the protein interaction of the WT cystinosin and different mutations. Furthermore, we focused on an atypical mutation concerning protein glycolsilation, ΔITILELP, that sometimes leads to severe forms.
Methodology & Theoretical Orientation: Co-immunoprecipitation and dynamic SILAC were used. All analyses were performed on a nanoRSLC Q-Exactive Plus MS.
Findings: We found cystinosin interacts with almost all components of vacuolar H(+)-ATPase and the Ragulator complex and with the small GTPases Ras-related GTP-binding protein A (RagA) and RagC. These interactions are lost when cystinosin carries severe loss-of-activity mutations. We also showed that wild-type cystinosin is very stable, while ΔITILELP is degraded three times more rapidly. We observed that in the lysosome, ΔITILELP is still capable of interacting with the V-ATPase complex and some members of the mTOR pathway, similar to the wild-type protein. Our interactomic and immunofluorescence studies showed that ΔITILELP is partially retained at the endoplasmic reticulum (ER).
Conclusion & Significance: Our results show a dual role for cystinosin as a cystine transporter and as a component of the mTORC1 pathway, and provide an explanation for the appearance of Fanconi syndrome in cystinosis. We also found that the high turnover of ΔITILELP, due to its immature glycosylation state in combination with low transport activity, might be responsible for the phenotype observed in some patients.
Recent Publications
1. Nevo N (2017) Impact of cystinosin mutations on protein stability by differential dynamic SILAC. Molecular and Cellular Proteomics (In press).
2. Lipecka J (2016) Sensitivity of mass spectrometry analysis depends on the shape of the filtration unit used for filter aided sample preparation (FASP). Proteomics. 16(13):1852-7.
3. Chhuon C (2016) Changes in lipid raft proteome upon TNF-α stimulation of cystic fibrosis cells. J. Proteomics. 145:246-53.
4. Andrzejewska Z (2016) Cystinosin is a component of the vacuolar H+-ATPase-Ragulator-Rag complex controlling mammalian target of rapamycin complex 1 signaling. J Am Soc Nephrol. 27(6):1678-88.
5. Ramond E (2015) Importance of host cell arginine uptake in Francisella phagosomal escape and ribosomal protein amounts. Mol Cell Proteomics. 14(4):870-81.
Marcus Krüger
University of Cologne, Germany
Title: Dynamic changes in the skeletal muscle proteome during denervation-induced atrophy
Time : 12:05-12:25

Biography:
Marcus Krüger studies Chemistry and received his PhD in Natural Sciences from the Univeristy of Halle-Wittenberg, working on basic-helix-loop-helix transcription factors in mouse. He did his Postdoctoral work at the Syddansk University in Odense working on quantitative mass spectrometry and stable isotope labeling of living animals. Since moving to the MPI for Heart and Lung Research in 2008, he became a Service Group Leader and in 2014 he was recruited as a Principle Investigator to the Institute for Genetics in Cologne. His current research interests includes quantitative mass spectrometryand the enrichment of posttranslational modifications in both health and disease related conditons.
Abstract:
Loss of neuronal stimulation enhances protein breakdown and reduces protein synthesis, causing rapid muscle mass loss. To elucidate the pathophysiological adaptations that occur in atrophying muscles, we used stable isotope labelling and mass spectrometry to accurately quantify protein expression changes during denervation-induced atrophy after sciatic nerve section in skeletal muscle. Additionally, mice were fed a SILAC diet containing 13C6 lysine for four, seven, or eleven days to calculate relative levels of protein synthesis in denervated and control muscles. Ubiquitin remnant peptides (K-e-GG) were profiled by immunoaffinity enrichment to identify potential substrates of the ubiquitin proteasomal pathway. Besides a protein expression profiling we used a pulse-SILAC labelling approach to identify differential Lys6 incorporation rates between control and denervated muscle. Enrichment of diglycine remnants identified 2100 endogenous ubiquitination sites and revealed a metabolic and myofibrillar protein diglycine signature, including myosin heavy chains (MyHC), myomesins and titin, during denervation. Comparative analysis of these proteomic datasets with known atrogenes using a random forest approach identified 92 proteins subject to atrogene-like regulation that have not previously been directly associated with denervation-induced atrophy. Comparison of protein synthesis and proteomic data indicated upregulation of specific proteins in response to denervation is mainly achieved by protein stabilization. This study provides the first integrated analysis of protein expression, synthesis and ubiquitin signatures during muscular atrophy in a living animal.
Jean-Michel Camadro
Institut Jacques Monod, France
Title: High-sensitivity, high-resolution MS-based quantitative proteomics by manipulation of protein isotope
Time : 12:25-12:45

Biography:
Abstract:
Many quantitative proteomics strategies rely on in vivo metabolic incorporation into proteins of amino acids with modified stable isotopes profiles. These methods give rise to multiple ions for each peptide, with possible distortion of the isotopologs distribution, making the overall analytical process complex. By reducing the isotopic composition of proteins in vivo, we bring a new dimension to in-depth, high resolution MS-based quantitative proteomics that alleviates these problems. We used U-[12C]-glucose as the metabolic precursor of all amino acids in yeast. This substantially increased the peptide monoisotopic ion intensity in bottom-up analyses, greatly improving identification scores and protein sequence coverage. Multiplexing samples of 12C composition varying from natural abundance to 100% 12C makes it possible to address quantitative proteomics, keeping all the critical information within a single isotopologs cluster. We applied this method to measure for the first time protein turnover at the proteome scale in the pathogenic yeast Candida albicans.
Bin Huang
Kaohsiung Medical University, Taiwan
Title: Application of embryogenic mouse as a platform in identifying human missing proteins
Time : 12:45-13:05

Biography:
Bin Huang gained his PhD degree from Department of Plant Science, National Taiwan University. He was also trained in Cardiology during Postdoctoral Fellow. Now he has the expertise in gaseous molecules-mediated post-translational proteome, particular for NO-mediated S-nitrosylation in the vascular system, and also the behaviors of mitochondrial fusion/Fission that evaluating cell aging and cancer cell drug-resistance. In addition to general research interests, he also has an administrative duty as a Vice Chief of Center for Stem Cell Research of Kaohsiung Medical University.
Abstract:
Missing proteins are those genes with detectable transcriptional mRNA while missed in detecting translational proteins either by mass spectrometry or Western blot. To date, missing proteins are thought as a “KEY” in triggering many early physiological responses and then disappear after these signaling cascades are turned on. That’s why missing proteins are always “MISSED” in numerous studies. Embryo contains many definitive genes to temporarily control subsequent organogenesis. Since the decoding of human and mouse whole genome, their proteins share high similarity in amino acid sequence. The embryos taken from pregnant mice for 6.5, 7, 7.5 and 8 days, the time for the onset of neuron plate formation, were analyzed by mass spectrometry. There were 17 missing proteins identified. The Western blot showed that NKX1 (Chr#4), PTCHD1 (Chr#X), OTOGL (Chr#12) and ASXL3 (Chr#20) were increased in day 7 and 7.5, while decreased in day 8. This transient expression indicates an association of missing proteins in neuron development. As a result, the embryonic mice could be the most suitable platform for detecting missing proteins and elucidating their role in organogenesis.
Alexander I Archakov
Institute of Biomedical Chemistry, Russia
Title: Sensitivity, specificity and accuracy of the targeted (SRM) and shotgun (panoramic) mass-spectrometry technologies
Time : 14:05-14:25
Biography:
Archakov A I is a Full Member of the RAS, Professor and Scientific Advisor of Institute of Biomedical Chemistry. He had organized a scientific school to study molecular organization and functioning of oxygenase cytochrome P450-containing systems, molecular mechanisms of the structure and function of membranes and biological oxidation. Under his guidance, the institute’s members have developed a fundamentally new pharmaceutical composition “Phosphogliv” with antiviral activity for the treatment of liver diseases of various etiology. His present-day/current areas of expertise relate to research in the field of post-genomic technologies, nanobiotechnologies, proteomics, development of approaches to create personalized medicine of the future. He is the pioneer in the development of proteomics in Russia. Currently, he is the international “Human proteome” Project Coordinator in Russia/the coordinator representing Russia in the international “HP” project. He is one of the Russia’s top 100 scientists by Hirsch number-27. He is the author of more than 700 scientific works including about 482 scientific articles, 6 monographs, 30 patents and author’s certificates. He was Scientific Adviser for 15 Doctor students and more than 60 PhD theses. He is a winner of three State Prizes of the USSR, the RSFSR and of the Russian Federation, the Russian Federation Government Award, the Bach Prize of the USSR Academy, the Order "For Merit to the Fatherland” (IV, III, II class).
Abstract:
There are about 2,579 missing proteins (NeXtProt PE2,3,4) in the human proteome. They have not been detected in any biological sample using existing experimental methods. They may be not translated, or presented in low-copied state, so existing methods could not be used to detect them. In this work, we compared existing proteomic mass spectromic technologies by using UPS2 set as control (Sigma-Aldrich) and Chromosome 18 encoded proteins detected in human liver and HepG2 cells. Targeted (Selected reaction monitoring, SRM) and panoramic (Shotgun LC-MS/MS) mass spectrometric methods were compared by sensitivity, specificity and accuracy similarly to the FDA diagnostic methods. Transcriptomic analysis of the same sample of human liver and HepG2 cells, or calibration standard UPS2 set was used as a “golden” standard. Proteomic analysis results were compared with the golden standard for true and false identifications revealing. Mass spectromic methods were evaluated using FDA indicators. Sensitivity of SRM in pure UPS2 solution is 92% (44 proteins from 48 detected), and in biological matrix (E. coli extract or human blood plasma) it decreases to 63%. Shotgun LC-MS/MS reveals 23 proteins in the pure UPS2 solution and 11 proteins in E. coli extract. In HepG2 cell line and liver tissue shotgun LC-MS/MS demonstrated sensitivity 6%, and SRM - 35% with the transcriptome “golden” standard. Both methods have high specificity (more than 90%), but the accuracy is only 57% for SRM and 19% for shotgun. Using indicators of sensitivity, specificity and accuracy for proteomic methods demonstrated, that proteins are “missing” in the sample due to different reasons. For example, chemical noise from other molecules in the biological matrix may interfere signal/noise ratio. As a result, MS-signals from presenting proteins may be lost or new MS-signals may appear that is the reason of false positive results. Thus, biological matrix significantly affects the list of detected proteins, which are existing in the mixture in low-copied state (≤10-9M, 109 copies in 1Ï»L).
Recent Publications
1. Zgoda V G et al. (2013) Chromosome 18 transcriptome profiling and targeted proteome mapping in depleted plasma, liver tissue and HepG2 cells. Journal of Proteome Research. 12(1): 123–134.
2. Ponomarenko E A et al. (2014) Chromosome 18 transcriptoproteome of liver tissue and HepG2 cells and targeted proteome mapping in depleted plasma. Journal of Proteome Research. 13(1): 183–190.
3. Poverennaya E V et al. (2016) State of the art on chromosome 18-centric HPP in 2016: transcriptome and proteome profiling of liver tissue and HepG2 cells. Journal of Proteome Research. 15(11): 4030–4038.
Theo Crosson
Proteintech Group Europe, UK
Title: Fusion protein raised antibodies for evaluating mass spectrometry results
Time : 14:25-14:45
Biography:
Theo Crosson is the account manager for France territory at Proteintech. Theo has studied biology and biotechnology at the ENSTBB in Bordeaux where he got an engineering degree. He has worked in research in Next Generation sequencing for a year at New England biolabs. He then completed his education with a specialized master in biotech management at the ESC Grenoble. He is now working for Proteintech, a company committed to providing high-quality antibodies produced and validated in-house, with the aim of assisting French researchers in their use of antibodies.
Abstract:
Proteomics studies often rely on the use of antibodies. Their ability to bind any protein makes them powerful tools of use for quantification, detection, and characterisation of proteins. In particular, antibodies have been increasingly used in these past years for mass spectometry applications. Proteintech, founded by a group of scientists in 2002, is committed to manufacturing high quality antibodies in-house. Using whole-protein approach, longer time of immunization, and stringent validation procedures, Proteintech has been able to support research in proteomics. Its antibodies have been used for publications involving mass cytometry, IP-MS, and target evaluation from mass spectometry.
Wataru Aoki
Kyoto Univeristy, Japan
Title: Mixed proteome analysis to elucidate complex symbiotic interactions
Time : 14:45-15:05

Biography:
Wataru Aoki obtained his PhD degree in Biology from Kyoto University in 2013. He worked in Osaka University as Research Fellow of the Japan Society for the Promotion of Science. He is now working in Kyoto University as an Assistant Professor. His current research interests include development of technologies for next generation proteomics.
Abstract:
Proteome analysis using LC-MS/MS is an important approach for a comprehensive characterization of complex biological systems. Steady improvement in MS has transformed the depth of proteome analysis. However, the separative power of the techniques currently available is still insufficient to analyze complex proteome samples. To overcome this weak point, we have developed a monolithic super-long silica column with Dr. Minakuchi (Kyoto Monotech, http://www.k-monotech.co.jp/). The column showed very high separative performance, and LC-MS/MS system equipped with a monolithic column can identify many proteins in living cells in one-shot. Using this system, we developed a method of “mixed and quantitative proteome analysis” in which proteome samples from several different organisms were simultaneously analyzed without the need to isolate the individual living cells. Omitting the individual cell isolation steps is important because these steps are known to alter the states of protein networks by causing various artifacts from unnecessary stresses. Our methods provide novel insights into the relationship between organisms with complex interactions and should lead to a better understanding of mechanisms of infectious diseases and symbiosis.
- Bioinformatics and Computational Biology | Biostatistics - An Approach to Bioinformatics | Immunology and Drug Discovery
Location: Paris, France
Session Introduction
Dharmendra K Yadav
Gachon University, South Korea
Title: Synthesis, biological evaluation and molecular simulation studies of new arylated benzo[h]quinolines
Time : 15:05-15:25

Biography:
Dharmendra K Yadav received his PhD degree from CSIR-Central Institute of Medicinal and Aromatic Plants, Lucknow, India, in 2013. During Doctoral research, he focused on the Molecular Modeling and Drug Design. After completion of PhD, he moved to Hanyang University Seoul, Korea for Postdoctoral Research and worked on QNTR. During this period he had performed his research in Molecular Modeling on nanoparticles specially using QNTR, etc. After completion, he moved to University of Delhi, and All India Institute of Medical Sciences, Jodhpur, India. He has published more than 38 research papers in reputed international journals with high impact factor and has one US patent. He is continuing his research in Computer-Aided Drug Design Dynamics Simulation of Biological Networks and Plasma Medicine, etc.
Abstract:
In this study, we have carried out Gaussian-based 3D-QSAR model against the target COX-2 with good statistical significance (R2training=0.86) and predictability (Q2training=0.66, Q2test=0.84). The 3D-QSAR includes steric, electrostatic, hydrophobic, and hydrogen bond acceptor field indicators, whereas the potential field contributions indicate that the steric and hydrophobic features of the molecules play an important role in governing their biological activity. The anti-cancer activity of the benzo[h]quinolines was evaluated on cultured human skin cancer (G361), lung cancer (H460), breast cancer (MCF7) and colon cancer (HCT116) cell lines. The inhibitory effect of these compounds on the cell growth was determined by the MTT assay. Among the synthesized compounds 3e, 3f, 3h and 3j showed potential cytotoxicity against these human cancer cell lines. Effect of active compounds on DNA oxidation and on expression of apoptosis related gene was studied. While their bioavailability/drug-likeness was predicted to be acceptable but requires future optimization. These findings reveal that benzo[h]quinolines act as anti-cancer agents by inducing oxidative stress-mediated DNA damage. Molecular simulation study was performed to find binding conformations and different bonding behaviors, in order to reveal the possible mechanism of action behind higher accumulation of active benzo[h]quinolines with β-tubulin.
Recent Publications
1. Yadav DK, Kumar S, Saloni, Singh H, Kim MH, Sharma P. Misra S, Khan F (2017) Molecular docking, QSAR and ADMET studies of withanolide analogs against breast cancer. Drug Design, Development and Therapy 11:1859-1870.
2. Yadav DK, Rai R, Kumar N, Singh S, Misra S, Sharma P, Shaw P, Pérez-Sánchez H, Mancera RL, Choi EH, Kim MH, Pratap R (2016). New arylated benzo[h]quinolines induce anti-cancer activity by oxidative stress-mediated DNA damage. Scientific reports 6:38128.
3. Yadav DK, Dhawan S, Chauhan A, Qidwai T, Sharma P, Bhakuni RS, Dhawan OP, Khan F (2014). QSAR and docking based semi-synthesis and in vivo evaluation of artemisinin derivatives for antimalarial activity. Current Drug Target 15(8):753-61.
4. Yadav DK, Ahmad I, Shukla A, Khan F, Negi AS, Gupta A (2014). QSAR and docking studies on Chalcone derivatives for anti-tubercular activity against M. tuberculosis H37Rv. Journal of Chemometrics 28: 499-507
5. Yadav DK, Kalani K, Singh AK, Khan F, Srivastava SK, Pant AB (2014). Design, synthesis and in vitro evaluation of 18β-glycyrrhetinic Acid derivatives for anticancer activity against human breast cancer cell line MCF-7. Curr Med Chem 21(9):1160-70.
Kathy Tzeng
Optum (UnitedHealth Group), USA
Title: From genomics to personalized healthcare
Time : 15:25-15:45

Biography:
Kathy Tzeng, PhD is a Distinguished Engineer in the Advanced Applied Technology team at Optum, with a focus on Genomics and Deep Learning. Prior to joining Optum, she was IBM’s worldwide Technical Team Lead for Healthcare and Life Science in the Systems group. Before joining IBM, she worked in Boehringer Ingelheim Pharmaceuticals, where she led several drug discovery projects in the area of Bioinformatics, Proteomics and Genomics. She received her PhD in Genetics from Iowa State University.
Abstract:
With the advent of Next Generation Sequencing (NGS) technologies that have significantly reduced the cost and time required to sequence human genomes, personalized healthcare is becoming a reality. Genomic information is increasingly an important determinant of clinical diagnosis and treatment. A bioinformatics challenge central to personalized healthcare is to identify the associations between genotypic and clinical observations, and to improve treatment outcomes by associating patients with known genomic specific treatments. Several analytics approaches and reference architecture to support high velocity software and large volume of unstructured data will be discussed
Recent Publications
1. Ogasawara T, Cheng Y, Tzeng TK (2016) Sam2Bam: High Performance Framework for NGS Data Preprocessing tools. PLOS ONE. https://doi.org/10.1371/journal.pone.0167100.
2. Sachdeva V, Kistler M, Tzeng TK (2010) Enabling Bioinformatics Algorithm on the CELL BE Processor. Chapter VII in Book “Scientific Computing with Multicore and Accelerators” CRC Press.
3. Sachdeva V, Kistler M, Speight E, Tzeng TK (2008) Exploring the viability of the Cell Broadband Engine for bioinformatics applications. Parallel Computing. 34(11): 616-626.
Mari van Reenen
North-West University, South Africa
Title: XERp a novel approach to variable selection for classification
Time : 16:05-16:25

Biography:
Mari van Reenen is Head of Bioinformatics at the Centre for Human Metabolomics, North-West University (Potchefstroom Campus), South Africa as well as a PhD candidate at the Department of Statistics, Faculty of Natural Sciences North-West University (Potchefstroom Campus), South Africa. The new statistical methods presented here form part of her PhD study aimed at developing new statistical approaches that can account for the nuances of metabolomics data. Her continued work and research aims to bridge the gap between the assumptions statistical tools often require and the reality of experimental work, from experimental design to data analysis.
Abstract:
Statement of the Problem: Variable selection and classification can become complex due to the large number and sources of missing values often present in data, specifically data generated in GC-MS based metabolomics experiments. Missing values are set to zero under certain conditions resulting in a mixture distribution which are difficult for most statistical approaches to account for.
Methodology: ERp is a variable selection and classification method based on minimized classification error rates, from a control and experimental group. ERp tests the null hypothesis that there is no difference between the distributions of the two groups. Significant variables are can discriminate between the two groups and provide insight into the biological mechanisms driving group differences. XERp is an extension of ERp that takes zero-inflated data into account. XERp addresses two sources of zero-valued observations: (i) zeros reflecting the complete absence of a metabolite from a sample (true zeros); and (ii) zeros reflecting a measurement below the detection limit.
Findings: XERp performs well with regard to bias and power. XERp was also applied to a GC-MS dataset from a metabolomics study on tuberculosis meningitis in infants and children and generated a list of discriminatory variables which informed the biological interpretation of the data. XERp also attained satisfactory classification accuracy for previously unseen cases in a leave-one-out cross-validation context.
Conclusion & Significance: XERp is able to identify variables that discriminate between two groups by simultaneously extracting information from the difference in the proportion of zeros and shifts in the distributions of the non-zero observations. XERp uses simple rules to classify new subjects and a weight pair to adjust for unequal sample sizes or sensitivity and specificity requirements.
Recent Publications
1. Van Reenen, M Westerhuis J A, Reinecke C J and Venter J H (2017) Metabolomics variable selection and classification in the presence of observations below the detection limit using an extension of ERp. BMC Bioinformatics, 18(1):83.
2. Irwin C, van Reenen M, Mason S, Mienie L J, Westerhuis J A, and Reinecke C J (2016) Contribution towards a metabolite profile of the detoxification of benzoic acid through glycine conjugation: an intervention study. PloS one. 11(12): e0167309.
3. Van Reenen M Reinecke C J, Westerhuis J A and Venter J H (2016) Variable selection for binary classification using error rate p-values applied to metabolomics data. BMC Bioinformatics. 17(1):33.
4. Moutloatse G P, Bunders M J, van Reenen M, Mason S Kuijpers, T W Engelke U F and Reinecke C J (2016) Metabolic risks at birth of neonates exposed in utero to HIV-antiretroviral therapy relative to unexposed neonates: an NMR metabolomics study of cord blood. Metabolomics. 12(11):175.
5. Mason S, van Furth A M T, Solomons R, Wevers R A, van Reenen M and Reinecke C J (2016) A putative urinary biosignature for diagnosis and follow-up of tuberculous meningitis in children: outcome of a metabolomics study disclosing host–pathogen responses. Metabolomics, 12(7):1-16.
Saloni
Gachon University, South Korea
Title: Molecular dynamics simulations and ADME studies of benzopyran class of selective COX-2 inhibitors for inflammatory activity
Time : 16:25-16:45

Biography:
Saloni received her Post-graduation in Applied Chemistry from Amity University, Noida, UP, India in 2016. During her Post-graduation, she completed her summer training at the 'Centre for Aromatic Plants', Uttarakhand, India where she got brief knowledge of the different medicinal properties of various aromatic plants and also learned to use few instruments in the lab. She also worked as the Research Trainee in the Dept. of Chemistry, University of Delhi, India. She is presently working as a PhD student in College of Pharmacy, Gachon University, Incheon city, Korea. She has published two research articles in reputed international journals with high impact factor. She is continuing her research in Computer-Aided Drug Design Dynamics Simulation of Biological Networks and Plasma Medicine, etc.
Abstract:
In the present work, 3D-QSAR model was derived by partial least squares method for the prediction of anti-inflammatory activity of benzopyran class of compounds against the COX-2 (cyclooxygenase-2). Partial least squares showed high correlation significant model with (R2training=0.866) and predictability (Q2training=0.66) and indicated that physiochemical descriptors namely, steric, electrostatic, hydrophobic, and hydrogen bond acceptor field indicators, correlate well with activity, whereas the potential field contributions indicate that the steric and hydrophobic features of the molecules play an important role in governing their biological activity. A molecular docking interaction pattern analysis reveals the importance of Tyr-361 and Ser-516 of the COX-2 active site for X-ray crystal structures and this class of molecules. Thus the molecular modeling based approaches provided an improved understanding in the interaction between benzopyran class and COX-2 inhibition. These findings may be of immense importance in the anti-inflammatory drug development of an inexpensive and benzopyran class of compounds.
Recent Publications
1. Yadav DK, Kumar S, Saloni, Singh H, Kim MH, Sharma P Misra S, Khan F (2017) Molecular docking, QSAR and ADMET studies of withanolide analogs against breast cancer. Drug Design, Development and Therapy 11:1859-1870.
2. Yadav DK, Rai R, Kumar N, Singh S, Misra S, Sharma P, Shaw P, Pérez-Sánchez H, Mancera RL, Choi EH, Kim MH, Pratap R (2016) New arylated benzo[h]quinolines induce anti-cancer activity by oxidative stress-mediated DNA damage. Scientific reports 6:38128.
3. Yadav DK, Dhawan S, Chauhan A, Qidwai T, Sharma P, Bhakuni RS, Dhawan OP, Khan F (2014) QSAR and docking based semi-synthesis and in vivo evaluation of artemisinin derivatives for antimalarial activity. Current Drug Target 15(8):753-61.
4.Yadav DK, Ahmad I, Shukla A, Khan F, Negi AS, Gupta A (2014) QSAR and docking studies on Chalcone derivatives for anti-tubercular activity against M. tuberculosis H37Rv. Journal of Chemometrics 28: 499-507
5. Yadav DK, Kalani K, Singh AK, Khan F, Srivastava SK, Pant AB (2014) Design, synthesis and in vitro evaluation of 18β-glycyrrhetinic Acid derivatives for anticancer activity against human breast cancer cell line MCF-7. Curr Med Chem 21(9):1160-70.
Fatemeh Koddam
Islamic Azad University, Iran
Title: Molecular Dynamics based screening for Cag A protein: Using Quercus brantii extract substances as lead-like
Time : 16:45:17:05

Biography:
Fatemeh Khoddam is a young researcher at Azad University of Tehran. Her main research interests are bioinformatics and drug design. Fatemeh studied Biophysics (Bachelor of Science) and then complete her education in Microbiology (Master of Science) both at Azad University. After spending a term in Bioinformatics in November 2014 she started her research in a team for inhibiting Listeria disease. During her first team work experience, Fatemeh started teaching molecular biology in a private institute in Tehran and she could establish a Drug Design Department in February 2016. Fatemeh is planning to apply for PHD in Drug Design.
Abstract:
Statement of the Problem: Gastric cancer is among the fifth common malignancy and third cause of cancer related mortality worldwide (approximately 700000 victims registered each year). Helicobacter pylori, that has been closely related to gastric ulcers and adenocarcinoma, infects nearly more than 50% of the entire human population based on presence or absence of CagA gene-encoded CagA protein, H. pylori divided into 2 strains: CagA positive and CagA negative. Currently, it has been demonstrated that CagA positive strain directly effects on gastric cancer incidence, so that H. pylori infection may lead to gastric cancer if it is getting chronic. But due to antibiotics resistance module which revealed by H. pylori strains, significance of discovering new generation of antibiotics is certainly tangible.
Methodology & Theoretical: In this study Quercus brantii as an Iranian aborigine, which rich extract called Shookeh has been obtained from it, used whereas leading like compound to inhibit specifically Cag A protein. since utilization of GC MASS technique for analysis Shookeh, Furfural was identified as the most abundant compound. The structure of Cag A protein with PDB ID 4IRV used for expectancy of binding affinity between the protein and Furfural by PyRx autodock vina software. Whereof the aim of this study is to present an efficient drug-like substance we respectively engineered furfural by Hyperchem software and make carbon nano tube around substance by SAMSON software to making an improved inhibitor for Cag A protein.
Conclusion & Significance: Molecular docking analysis indicates -4.3 kcal/mol for binding affinity and pharmacokinetic analysis by using FAFDrugs4 server does not predict any oral toxicity for furfural. After engineering the substance binding affinity reduces up to -6.8 kcal/mol with no oral toxicity even with carbon nanotube.
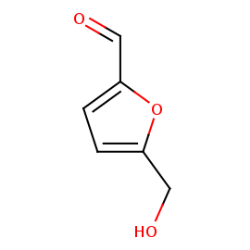
Figure 1: Furfural substance obtained from pubchem NCBI and 3D structure of Cag A protein
Recent Publications
1. HATAKEYAMA, Masanori (2017) Structure and function of Helicobacter pylori CagA, the first-identified bacterial protein involved in human cancer. Proceedings of the Japan Academy, Series B
2. Lai, Chih-Ho et al (2011) Helicobacter pyloriCagA-mediated IL-8 induction in gastric epithelial cells is cholesterol-dependent and requires the C-terminal tyrosine phosphorylation-containing domain. FEMS microbiology letters.
3. Mishra, Ajay Kumar (2013) Nanomedicine for drug delivery and therapeutics, John Wiley & Sons.
4. Shrivastava, Arpit Kumar et al (2017) Insilico identification and validation of a novel hypothetical protein in Cryptosporidium hominis and virtual screening of inhibitors as therapeutics. Parasitology research.
5. Murata-Kamiya, Naoko (2010) Helicobacter pylori exploits host membrane phosphatidylserine for delivery, localization, and pathophysiological action of the CagA oncoprotein. Cell host & microbe.