Day 2 :
Keynote Forum
Judit Ovádi
Hungarian Academy of Sciences, Hungary
Keynote: Challenging drug target for moonlighting and chameleon proteins
Time : 09:30-10:00

Biography:
Judit Ovádi has expertise in Biochemistry, Enzymology and Molecular Biology. She defined the metabolic channeling at microscopic and macroscopic levels as a powerful mechanism to control and direct metabolic pathway at crossroads. Her research team showed the sensing potency of the microtubule network at system level that can regulate signaling pathways due to its decoration by interacting partners. They discovered a unique brain-specific protein denoted Tubulin Polymerization Promoting Protein (TPPP/p25) that displays two exciting characteristics: intrinsically unstructured and enriched in brain inclusion in the case of Parkinson disease and other synucleinopathies. The structural and functional features of this protein have been characterized at different levels of organizations under physiological and pathological conditions. She has supervised several DSc and PhD dissertations. She was an invited Visiting Professor in USA, Spain and Italy; Invited Speaker in international congresses in the last few years in Tokyo, San Francisco, Jerusalem, Orleans, Nice. At present, she is a Professor Emerita, Laureata Academiae of the Research Center of Hungarian Academy of Sciences.
Abstract:
The conformational diseases such as Parkinson’s disease (PD) and Multiple System Atrophy (MSA) represent an important group of neurodegenerative disorders. The hallmarks of these diseases are the a-synuclein (SYN) and the recently discovered Tubulin Polymerization Promoting Protein (TPPP/p25). Both proteins are disordered with chameleon characteristics and expressed distinctly in neurons and oligodendrocytes (OLGs), respectively; notwithstanding they are co-enriched and co-localized in pathological inclusions in the case of PD and MSA. TPPP/p25 is the prototype of the Neomorphic Moonlighting Proteins by displaying both physiological and pathological functions due to their interactions with distinct partners. At physiological conditions TPPP/p25 modulates the dynamics and stability of the microtubule system; its expression is crucial for the differentiation of OLGs, the major constituents of the myelin sheath. The assembly of TPPP/p25 and SYN, as fatal initiative, of the etiology of PD and MSA has been established. Due to the unique structural and functional features of TPPP/p25, a new innovative strategy has to be evaluated to inhibit and/or destruct specifically the interaction of TPPP/p25 with SYN; this could be fulfill by targeting of the interface of the pathological complex without affecting the physiological one. Our studies underline that targeting multifunctional proteins is a challenging task; nevertheless, the validation of a drug target can be achieved by identifying the interface of complexes of the partner proteins existing at the given pathological conditions.
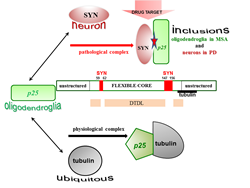
Figure 1: Distinct interfaces of TPPP/p25 are involved in its hetero-association with tubulin and SYN as physiological and pathological partners. The interaction of the disordered TPPP/p25 with tubulin results in significant conformational changes; this effect does not manifest itself in the case of its association with SYN. Although the deletions within the middle CORE segments do not abolish the binding of TPPP/p25 mutants to SYN, except in the case of the double truncated double loop mutant (DTDL), due to its chameleon feature; however, a potential segments (marked red) were identified as potential drug target.
Keynote Forum
Jarrod A Marto
Harvard Medical School, USA
Keynote: CITe-Id as a novel chemoproteomic platform to characterize covalent kinase inhibitors
Time : 10:00-10:30

Biography:
Jarrod A Marto is internationally recognized for his expertise in the development and use of state-of-the-art mass spectrometry and other bioanalytical techniques to characterize cellular communication pathways that underlie normal physiology and human diseases. His lab pursues technology development in mass spectrometry for quantitative analysis of primary human tissues or high-fidelity model systems. He has published widely in the areas of basic chemistry, analytical science, advanced instrumentation, mass/bio-informatics, and cancer biology.
Abstract:
The therapeutic value of targeting protein kinases is demonstrated by the small molecule inhibitors receiving regulatory approval primarily for cancer therapy. Despite these successes, only a handful of truly selective inhibitors have been developed for the nearly 600 human kinases. The recent approval of cysteine-directed covalent inhibitors of BTK and EGFR has reignited interest in covalent kinase therapeutics. One advantage of covalent drugs is their ability to potently and permanently disable protein function often with only transient drug exposure. We are focused on probes which covalently modify members of the cys-kinome, the subset of approximately 200 kinase which harbor a targetable cysteine residue in proximity to the ATP-binding site. We have developed quantitative mass spectrometry approaches which enable site-level interrogation of proteins targeted by irreversible inhibitors on a proteome-wide scale. For individual probes which target kinases such as EGFR, JNK, BMX, FGFR, CDK7, or BTK, we typically identify several hundred intracellular protein targets. We developed a companion, competition-format assay called 'CITe-Id' which discriminates non-specific versus selective, concentration-dependent inhibitor binding to protein targets. Importantly we successfully differentiate the repertoire of binding targets for probes which comprise structurally similar analogs, suggesting an efficient mechanism for medicinal chemistry optimization of second-generation inhibitors. Finally, our quantitative approach provides important clues for development of inhibitors targeting obscure kinases. The combination of structure-guided synthesis informed by CITe-Id chemoformic target and site identification provides a scalable platform that delivers first-in-class covalent chemical probes that may serve as useful starting points for future small molecule therapeutics.
Keynote Forum
Magnus S Magnusson
University of Iceland, Iceland
Keynote: From protein to human cities the first and only large-brain mass-societies: Structural and functional
Time : 10:30-11:00

Biography:
Magnus S Magnusson is a Research Professor. He completed PhD in 1983 from the University of Copenhagen. He is an Author of the T-pattern model initially focused on the real-time organization of behavior has co-directed DNA analysis. He presented numerous papers and invited talks at international mathematical, neuroscience, proteomics, bioinformatics and science of religion conferences and at leading universities in Europe, USA and Japan. He is a Deputy Director during 1983-1988 at Anthropology Laboratory, Museum of Natural History, Paris and repeatedly invited temporary Professor in Psychology and Ethology (Biology of Behavior) at the University of Paris (V, VIII & XIII). Since 1991, he is the Founder and Director of the Human Behavior Laboratory, University of Iceland, in formalized collaboration between 28 European and American universities based on “Magnusson’s analytical model” initiated at University René Descartes Paris V, Sorbonne, in 1995.
Abstract:
Striking similarity, but no self-similarity, exist between the mass-societies (i.e., of approximately >104 individuals) of such distantly related organisms as social insects and modern humans, while striking self-similarity exists between their structured mass-societies and those of their citizens that are mass-societies of cells that again are mass-societies of proteins (also called Cell Cities). Natural, as opposed to mathematical, self-similarity should probably be expected in a generally (statistical pseudo) fractal universe. This may be exemplified by the statistically self-similar repeated pattern type, called T-Pattern that is also characterized by significant translation symmetry, as it has been detected among other in the dynamic behavior and interactions of humans and in networks of neurons in living brains, while resembling (spatial) patterns on DNA. The brain-less mass-societies of proteins may thus provide useful and even essential ideas for the understanding of the biologically recent and first and only large-brain mass-societies, mostly evolved at the speed of cultural evolution among brains biologically evolved during a far longer (nomadic and illiterate) small-group “everybody-knows-everybody” past. Structural and functional (self-)similarities between protein and human mass societies are explored among others in terms of T-patterns appearing across time and space and from protein to human mass-societies, where extensive copying and spreading of standard T-patterned strings (texts) existing and evolving outside brains, seem reflections of the social ways of proteins, much as human schools, laws, religions and money.
- Protein Expression and Analysis | Omics Data Integration and Databases | Mass Spectrometry in Proteome Research | Proteomics in Biochemistry and Molecular Biology
Location: Paris, France
Session Introduction
Marwa Eltoweissy
Georg August Medical University of Göttingen, Germany and Alexandria University, Egypt
Title: Short-time increase of glucose concentration in PDS results in extensive removal and high glycation level
Time : 11:20-11:40

Biography:
Marwa Eltoweissy has completed her PhD through a scholarship and cooperation work between faculty of Science, Alexandria University, Egypt and Rheinische Friedrich-Wilhelms-University Medical Center Bonn, Institute for Physiology II, Germany. She achieved Postdoctoral studies at the Gastroenterology and Endocrinology department, Georg-August University Medical Center, Göttingen, Germany. She received the Doctor of Natural Sciences degree through her work at the Nephrology and Rheumatology department, Georg-August University Medical Center, Göttingen, Germany. She worked as a major Scientific Researcher at the later department and is an Assistant Professor of Physiology at the Zoology department, Alexandria University, Egypt. She has published more than 30 papers in reputed journals and is serving as a reviewer for privileged journals. She has been involved in many international conferences and workshops as a speaker, member of Scientific Program Committees, Organizer, Session Chair/Co-chair and in conferences moderation. She is a Member of the Editorial Board of two journals in proteomics.
Abstract:
Background: Renal diseases constitute a major health risk in all societies. The prevalence of end-stage renal disease (ESRD) in adult European populations is above 10% with tendency to increase, posing a serious health threat. The treatment of ESRD involved the use of various dialysis procedures or kidney transplantation. Continuous ambulatory peritoneal dialysis (CAPD) uses the well flow peritoneum as a biological semipermeable membrane without an extra corporal blood circulation. Glucose solution is commonly used as dialysate compound. In our present study, we investigated the impact of short-time alteration of the glucose concentration and the osmolarity of the peritoneal dialysis solution (PDS) on protein removal.
Methods: Peritoneal dialysis liquids (PDL) were collected from 19 well-characterized CAPD patients treated with two types of PDS. The patients were subjected to short-time changes (4 h) of glucose concentration of PDS. The depletion of the six-interfering high abundant proteins from the PDL samples was performed with the Multiple Affinity Removal LC Column-Human 6. The resulting protein fractions were analyzed by 2D gel electrophoresis, differential in gel electrophoresis, mass spectrometry and 2D western blot.
Results: Proteomics investigation of the PDL fractions after depletion allowed the identification of 198 polypeptides, which equate to 48 non-redundant proteins. Comparative analyses of 2D gel electrophoresis protein pattern revealed a clear correlation between protein removal, PDS glucose concentration and osmolarity. An increase for 4 h in the PDS osmolarity (with 43-51 mOsmol/L) resulted qualitatively in 18-23% more protein removal in PDL. Moreover, 2D western blot analyses of the protein glycation pattern showed that the short-time increase in PDS glucose concentration (45-50 mM) resulted in significant alteration of the advanced glycosylation end products (AGEs) pattern.
Conclusions: The data presented in this study shed light on the quality of the protein lose during CAPD due to the glucose concentration in used dialysate. Moreover, we could demonstrate that higher glucose concentration in dialysis solution results in increased AGEs.
Pavlina Dolashka
Bulgarian Academy of Sciences, Bulgaria
Title: Proteomics analysis of Alzheimer's and antitumor activity of glycoproteins against bladder carcinoma
Time : 11:40-12:00

Biography:
Dolashka P and her group has wide experience in the isolation, purification, and characterization of biologically active compounds. She has more than 130 publications on these topics, 3 book chapters and 6 patents. She is Editor-in-board of 3 journals and representative IUPAC. She is coordinating several international research projects, sponsored by NATO (Brussels), the European Commission, Germany (DFG and BMBF), CNR (Italy), FWO (Belgium), China, and Ukraine.
Abstract:
Alzheimer's disease (AD) is the most common form of dementia. It is the sixth leading cause of death, and affects nearly 30 million people worldwide. Scopolamine and streptozotocin are widely utilized in chemically-induced dementia animal models to mimic specific pathophysiological pathways thought to underline AD. To the best of our knowledge, there is no report describing proteome analysis on scopolamine or streptozotocin AD animal models. Therefore, we conducted a comparative proteome analysis on CSF isolated from rats with chemically-induced dementia with the purpose of identifying protein biomarkers. Rodents were divided into three groups: rats with scopolamine-induced dementia, rats with streptozotocin-induced dementia and healthy controls. Proteins and peptides were separated from the isolated CSF into four fractions. Two low molecular peptide fractions, with mass below 3 kDa, and another with mass ranging from 3 to10 kDa were analyzed by mass spectrometry, while two other protein fractions, with mass between 10 and 50 kDa, and with mass higher than 50 kDa, were characterized by 2D-PAGE and the results were compared. The impact of hemocyanin on tumor cells was investigated by 2D-gel PAGE and several proteins showed indeed altered abundancies. The most effective inhibition of tumor cells is probably caused by a specific novel and unusual N-glycan oligosaccharide structure on HlH with methylated hexoses, an internal fucose residue connecting one GalNAc(ß1-2) and one hexuronic acid.
Theresia Conrad
Hans-Knöll-Institute, Germany
Title: Integration of multilevel OMICs data based on the identification of regulatory modules
Time : 12:00-12:20

Biography:
T Conrad studied bioinformatics at the Friedrich Schiller University Jena, Germany. During her studies, she spent several years at the Centre for Innovation Competence Septomics of the Jena University Hospital and Friedrich Schiller University focusing on the exploration of septic infections in consideration of the PIRO-concept. In 2015, she was awarded a Jena School for Microbial Communication (JSMC) fellowship and started her PhD in Bioinformatics at the Leibniz Institute for Natural Product Research and Infection Biology - Hans Knöll Institute. Her research focuses on developing multilevel models to obtain a deeper understanding of host-pathogen-interactions during fungal infections.
Abstract:
Complex scientific experiments provide researchers with a wealth of data from heterogeneous sources. Analyzed in its entirety, omics data provide a deep insight into the overall cellular processes of organisms. However, the integration of data from different cellular levels is challenging. Thus, there is a need for approaches dealing with this issue and in this study, we make use of transcriptome, proteome and secretome data from the human pathogenic fungus Aspergillus fumigatus challenged with caspofungin. Caspofungin is an antifungal drug targeting the fungal cell wall leading to a compensatory stress response. We analyze the experimental data based on two different approaches: first, we apply a simple approach based on the comparison of differentially regulated genes and proteins; secondly, we compare the cellular levels based on the identification of regulatory modules from protein-protein interaction networks. Our results show that both approaches associate the fungal caspofungin response with biological pathways like cell wall biosynthesis and carbohydrate metabolism. Compared to results of the simple approach, the regulatory modules show a notably higher consistency between the levels. The additional structural information of the networks provided by the module-based approach allows for topological analysis and the analysis of the temporal evolution of response. However, the quality of the module-based results depends on the comprehensiveness of the underlying protein-protein interaction network itself. Thus, while our results highlight the benefits and potential of a module-based analysis of multilevel omics data, future studies will have to focus on the expansion of organism specific protein-protein interaction networks.
Yinghong Pan
The National Key Facility for Crop Gene Resources and Genetic Improvement - ICS CAAS, China
Title: A proteomics strategy to analyze complex traits and gene functions
Time : 12:20-12:40

Biography:
Yinghong Pan worked for the Institute of Medicinal Plant Development (IMPLAD), Chinese Academy of Medical Sciences, and is engaged in research on medicinal plants and bioactive proteins during 1982-1999. Since 1999, he worked for the State Key Laboratory for Biology of Plant Diseases and Insect Pests (SKLBPI), Institute of Plant Protection, and the National Key Facility for Crop Gene Resources and Genetic Improvement (NFCRI), Institute of Crop Science, Chinese Academy of Agricultural Sciences, and he is engaged in research on plant proteomics. He has published more than 20 papers related to medicinal plants, and more than 55 related to proteins and proteomics.
Abstract:
Proteomics analysis of complex trait is useful to understand gene functions and trait mechanisms. But the reliability of proteomics analysis is affected by many factors, such as genetic background influences, sampling variations, and experimental conditions. In this strategy, four near isogenic lines (NILs) of dwarf male-sterile (DS) wheat with different genetic backgrounds were analyzed and a large mass spectrometry (MS) data from multiple batches were used to study the DS traits in wheat. At first, another and immature spike proteins from four NILs of DS wheat were prepared in different ways and detected with different mass spectrometry, and a total of 58170 protein groups were detected from sixteen independent experiments. Secondly the abundance distributions of proteins that detected at different frequencies and expressed types were analyzed. By using several simple formulae which were introduced to evaluate the reliability of protein expression, a database contained expression levels of 58170 protein groups and comprehensive evaluation values of 17187 proteins without duplications was established. As focusing on nuclear male sterile trait mechanism and the function of Taigu genie male-sterile wheat (TGMSW) gene ms2 in those NILs, proteomes of immature spike from three DS wheat NILs were analyzed under same conditions, and 160 differentially expressed proteins and 43 highly expressed proteins detected in this experiment were compared with database. Finally, it is determined that 28 proteins were closely related to male-sterile trait. The result show that large MS data from multiple batches is helpful for studying the complex traits and gene functions.
Leonora V Autus-Geniston
United Bayanihan Foundation and St. Paul University-Quezon City, Philippines
Title: STRING v9.1: Edges and nodes provide global functional association to protein crosstalk and integration
Time : 12:40-13:00

Biography:
Leonora V Autus-Geniston has completed his PhD from University of Santo Tomas. She is the Officer of United Bayanihan Foundation and an Assistant Professor at Saint Paul University–Quezon City, Philippines. He has published several papers in reputed journals on proteomics and pharmacogenomics, specifically on SNPs on drug metabolizing enzymes among Filipinos. She received the Best Poster Award in Health Science from National Academy of Science and Technology (Philippines) on “Method Development of nanoLC-MS/MS for Identification of Protein Biomarkers in the Urinary Proteomes of Prostate Cancer” and Best Poster in Penang, Malaysia on “Protective Effects of Thiamin on Genotoxicity in Mice”.
Abstract:
Prostate cancer was the number one cause of cancer in men in Asia. Urine like blood has a similar source of proteins making it an ideal matrix for the study. The research aims to identify proteins in the urinary proteomes of normal controls, benign prostate hyperplasia (BPH) and prostate cancer by initial separation using SDS-PAGE followed by nanoLC-Orbitrap MS/MS aided by ProteinProspector and XCalibur. STRING v9.1 predicted their functional protein association networks among the gene lists. Three hub proteins (B2M, GSN, IGHG1) and FN1 were seen in normal state and BPH, respectively. Six functional modules in normal controls and BPH were seen, and 12 in prostate cancer. Five and 15 unique proteins were seen in normal and BPH. HBA1, HBB and TTR were the distinct proteins observed in prostate cancer. A unique BPH module emerged from the normal state when their networks were overlapped. The modules at the center of the prostate cancer networks were tight and multiply connected with loosely-arranged periphery. The protein markers of prostate cancer, HBA1-HBB edge in module 1 in the periphery, while TTR in module 6 at the center was connected by haptoglobin (HP) from module 5. Prostate carcinopathogenesis from the normal state involved three protein switches: GSN-FN1, B2M-IGHG1 and CTSB-CSTB edges independent from BPH pathogenesis. From these switches, the expression of the known cancer functional modules emerged that linked further to HBA1-HBB-TTR hub. The details of these events will be discussed. Thus, the present study provides a solid framework for further understanding of prostate pathocarcinogenesis.
Altijana Hromic
University of Graz, Austria
Title: Structural view on substrate specificity of dipeptidyl peptidase III (DPP III)
Time : 14:00-14:20

Biography:
Altijana Hromic has completed her Master’s studies in Biochemistry and Molecular Biomedicine 2013 from Graz University of Technology and is attending her PhD studies at the University of Graz. She has published more than 10 papers in reputed journals and has extended experience in industry projects.
Abstract:
Dipeptidyl peptidase III (DPP III), also known as enkephalinase B, belongs to family of M49 of zinc-dependent metallopeptidases and cleaves dipeptides sequentially from the N-terminus of numerous bioactive substrates. These peptides are produced in the body and have been shown to act as neurotransmitters by interacting with their cognate receptors. Some of them form the endogenous opioid peptide system, which modulates large numbers of motivational, sensory and cognitive functions. Moreover, they contribute to the regulation of diverse physiological functions like signal transduction, gastrointestinal motility, social behavior with the aspect to drug addiction and immune functions. Many pharmacological experiments showed the role of DPP III in pain modulation and recently implicated its involvement in oxidative stress response or in the regulation of blood pressure, which fostered higher interest in investigation of this enzyme. DPP III is found in different species including higher mammals, but has also been described in lower eukaryotes like the yeast Saccharomyces cerevisiae and in some bacterial species like Porphyromonas gingivalis and Bacteroides thetaiotamicron. It has been reported that DPP III acts as post proline cleaving enzyme. This leads to the conclusion that it can develop amino acid pool by cleaving proline containing peptides which are usually resistant to hydrolysis by other aminopeptidases. This presentation provides a general overview of dipeptidyl peptidases III from different organisms, their biochemical and structural properties.
Dariusz Rakus
University of Wrocław, Poland
Title: Proteomic and immunofluorescent evidence for and against the astrocyte-to-neuron lactate shuttle
Time : 14:20-14:40

Biography:
Dariusz Rakus is the Head of the Department of Molecular Physiology and Neurobiology, at University of Wroclaw. He has published more than 50 papers in reputed journals about multifunctional proteins in glycolysis and glyconeogenesis and in the field of physiology and biochemistry of muscle, and brain, and cancer tissues.
Abstract:
Lactate derived from astrocytic glycogen has been shown to play a decisive role in induction of neuronal plasticity by stimulating memory formation in hippocampi of young animals but inhibiting it in old animals. Pharmacological inhibition of glycogen turnover in young animals blocked the basal transmission and memory formation while it improved significantly the neuronal plasticity in hippocampi of aged animals. Here we show, using quantitative proteomics and immunofluorescent microscopy, that aging is associated with an increase of glycogen metabolism enzymes concentration and shift in their localization, from astrocytes to neurons. These changes are accompanied with reorganization of hippocampal energy metabolism which is manifested by elevated capacity of aging hippocampal neurons to oxidize glucose in glycolysis and decreased ability of their mitochondria to produce energy. Our observations suggest that astrocyte-to-neuron lactate shuttle may operate in young hippocampi, however, during aging, neurons became independent on astrocytic lactate and they may start to produce lactate from blood-derived glucose.
- Bioinformatics and Computational | Bioinformatics Algorithms & Databases
Location: Paris, France
Session Introduction
K. Venkateswara Swamy
Dr. D Y Patil Biotechnology and Bioinformatics Institute, India
Title: Molecular modeling, docking, dynamics and simulation of deguelin and its derivatives with cyclin D1 and cyclin E in cancer cell signaling pathway
Time : 14:40-15:00

Biography:
K Venkateswara Swamy is currently working as an Associate Professor. He was awarded Young Scientist Award by ICMPHP, George Washington University, USA in 2013. He is a Visiting Faculty at Jaffna University, Sri Lanka. He has received SERB-DST project under Young Scientist Scheme in the year 2016 and is a member in various academic committee of bioinformatics at Dr. D Y Patil Vidyapeeth, Pune, India. He has published 23 national and international research papers in reputed journals. He was invited for national workshop in bioinformatics as Key Note Speaker by various Universities and Institutes. He is expertise with Molecular Modeling, Docking, Simulations and Dynamics of cancer proteins. He has already submitted 32 theoretical protein models to Protein Model Data Base (PMDB).
Abstract:
Deguelin is a major active ingredient and principal component in several plants, Derris trifoliata Lour. (Leguminosae), Mundulea sericea (Leguminosae), Tephrosia vogelii Hook.f. (Leguminosae) and potential molecule to target cancer cell signaling pathway proteins. As a complex natural extract, deguelin interacts with various molecular targets to exert its anti-tumor properties at nanomolar levels. Deguelin induced cell apoptosis by blocking anti-apoptotic pathways, while inhibiting tumor cell propagation and malignant transformation through p27-cyclin-E-pRb-E2F1- cell cycle control and HIF-1αVEGF antiangiogenic pathways. Our research explores the deguelin and its derivatives interaction with crystal structure of cyclin D1 (PDB ID: 2W96) and cyclin E (PDB ID: 2AST) to understand the better molecular insights. Molecular modelling of ligands (deguelin and its derivatives) were carried out by Avogadro software till stable confirmation obtained. The partial charges for the ligands were assigned as per standard protocol for molecular docking. All docking simulation were performed with AutoDock Vina on workstation. Virtual screening was done for all docked molecules based on binding energy and hydrogen bonding affinity. Molecular dynamics (MD) and Simulation (10 ns and 12 ns for cyclin D1 and cyclin E1 respectively) was done using GROMACS 5.1.1 software to explore the interaction stability. All the stable confirmations for cyclin D1 and cyclin E proteins trajectories was captured at various time intervals. Few compounds screened based on high affinity as inhibitors for cyclin D1 and cyclin E and may inhibit the cell cycle in cancer cell signaling under in vitro and in vivo experiments.
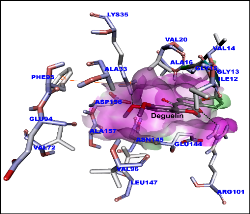
Figure 1: Molecular docking of deguelin with cyclin D1 protein. Surface shown in pink. Amino acids and deguelin hydrogen bond interaction with dotted lines.
Recent Publications
1. Prasad Dandawate, Kiranmayi Vemuri, K Venkateswara Swamy, Ejazuddin M Khan, Manjula Sritharan and Subhash Padhye (2014) Synthesis, characterization, molecular docking and anti-tubercular activity of Plumbagin-Isoniazid Analog and its β-cyclodextrin conjugate. Bioorganic & Medicinal Chemistry Letters 24(21):5070-5075.
2. Prasad Dandawate, Aamir Ahmad, Jyoti Deshpande, K Venkateswara Swamy, Ejazuddin M Khan, Madhukar Khetmalas, Subhash Padhye and Fazlul Sarkar (2014) Anticancer phytochemical analogs 37: Synthesis, characterization, molecular docking and cytotoxicity of novel plumbagin hydrazones against breast cancer cells. Bioorganic & Medicinal Chemistry Letters, 24:2900-2904.
3. P Sharma, P Patil, N Rao, K V Swamy, M B Khetmalas and G D Tandon (2014) Mapping biodiversity of indigenous freshwater chlorophytes. Research Journal of Pharmaceutical, Biological and Chemical Sciences 5(3):1632-1639.
4. Shibnath Ghataka, Alok Vyas, Suniti Misra, Paul O’Briend, Ajit Zambre, Victor M Fresco, Roger R Markwald, K Venkateshwara Swamy, Zahra Afrasiabif, Amitava Choudhury, Madhukar Khetmalas and Subhash Padhye (2013) Novel di-tertiary-butyl phenylhydrazones as dual cyclooxygenase-2/5-lipoxygenase inhibitors: synthesis, COX/LOX inhibition, molecular modeling, and insights into their cytotoxicities. Bioorganic & Medicinal Chemistry Letters 24:317-324.
5. Siddiqui A, Dandawate P, Rub R, Padhye S, Aphale S, Moghe A, Jagyasi A, Venkateswara Swamy K, Singh B, Chatterjee A, Ronghe A and Bhat H K (2013) Novel Aza-resveratrol analogs: Synthesis, characterization and anticancer activity against breast cancer cell lines. Bioorganic & Medicinal Chemistry Letters 23:635–640.
Sherief El Rweney
Royal Holloway, University of London
Title: Drug repositioning system using the power of network analysis and machine learning to predict new indications for the approved drugs “drug repositioning and rate the level of the drug similarityâ€
Time : 15:00-15:20

Biography:
Sherief El Rweney has over 13 years experience in computer science systems and information technology, he has done Master’s degree at Royal Holloway University of London in Computer Science, Data Analysis, Machine Learning, And Bioinformatics , the merging between his experience in bioinformatics and machine learning study motivated him to build the Drug Repositioning System based on protein interaction where can be used to predict new indication for the approved drugs and help drugs scientists to go easy through their drug discovery research. He is aiming to develop the Drug Repositioning System by merging many other factors beside the protein interaction to strength the predication of the drugs repositioning.
Abstract:
Drug discovery is a lengthy process, taking on average 12 years for the drugs to reach the market –but as Sir James Black OM once said, “the best way to discover a new drug is to start with the old one”. As result, this will drive to Drug repositioning concept. Drug Repurposing and repositioning is finding a new clinical use for an approved drug. There are many factors that can be used to predict new target disease. i.e. protein-protein interaction, chemical structure, gene expression and functional genomics, Phenotype and side effect, genetic variation and Machine learning. Protein-protein interaction (PPI) is Physical contacts with molecular docking between proteins that occur in a cell or in a living organism in vivo. There are Two Alternative Approaches PPI “Binary: yeast twoâ€hybrid (Y2H) and coâ€complex: (TAPâ€MS)”. Drug Repositioning System, is a system built based on protein-protein Binary interaction to predict new targets for the approved drugs. The system curates the data sets for human PPI, Drugs and diseases from well-known online sources (PPI from HRPD, drugs from DrugBank, Diseases from DisGeNET), Drug Repositioning System relates the 3 data sets based on genes name. Drug Repositioning System consisting of two interfaces: backend system where the curated data sets stored based on rational database and frontend web interface where the end users can use many search engines to search inside the system for diseases, genes and drugs to predict and find new targets for the approved drugs based on protein interactions, from the web interface the user can make analysis based on his search result and build network between the genes, diseases and drugs and generate statistics to be able to answer his question. There are many Questions that can be answered by Drug Repositioning System and generate statistics: for example the main question is can we find new indications for existing approved drugs. Drug similarity: from the Drug Repositioning System we can measure the percentage of drugs similarity between any pair genes interaction based on the number of shared drugs between them to rate the level of drug repositioning strength and then use the ROC ( receiver operating characteristic curve) analysis.
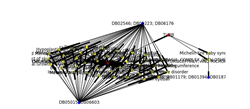
Figure 2: in the left side of the picture, it’s clear that there are two groups of drugs target numerous diseases related to HDAC6 gene, and also on the right side we will find one group of drugs targets two groups of disease that are related to TUBB gene while there link between TUBB and HDAC6 indicates the interaction between them. As result we can make drug repositioning between the two genes.