Day 17 :
- Track 1: Evolutionary & Structural Bioinformatics
Track 2: Emerging Trends in Proteomics & Genomics
Track 3: Frontiers of Systems Biology in Bioinformatics

Chair
Alexandru G Floares
SAIA-Solutions of Artificial Intelligence Applications, Romania

Co-Chair
Elena Papaleo
Danish Cancer Society Research Center, Denmark
Session Introduction
Wenzhong Xiao
Stanford University School of Medicine, USA
Title: Of men and not mice: Comparative genomic analysis of human diseases and mouse models
Time : 16:10-16:30

Biography:
Wenzhong Xiao directs the Computational Genomics lab at Stanford Genome Technology Center, Stanford Medical School and is responsible for solving computational problems in the development of new genomic and proteomic techniques and their applications. He is also the Director of the Immuno-Metabolic Computational Center at Massachusetts General Hospital. At the interface of computation, genomics and medicine, he focuses on “Computational challenges in development and application of omics for diagnosis, prevention and therapy, especially of human immune and metabolic diseases”.
Abstract:
A cornerstone of modern biomedical research is the use of mouse models to explore basic disease mechanisms, evaluate new therapeutic approaches and make decisions to carry new drug candidates forward into clinical trials. However, few of these human trials have shown success. Here, we systematically compared the genomic response from publically available datasets of patients of different acute inflammatory diseases and corresponding murine models; and showed that, although inflammation from different etiologies resulted in highly similar genomic responses in humans, the responses in mouse models correlated poorly with the human disease and also with one another. Among genes changed significantly in humans, the murine orthologs are close to random in matching their human counterparts. Our findings suggest that a disease model shall be carefully (re) examined to see how well it reproduces the human disease at the molecular level because virtually every drug and drug candidate target gene product(s). In addition, our comparisons of trauma patients reveal genomic signature between complicated and uncomplicated outcomes, and specific mediators which serve as predictive biomarkers for the development of targeted treatments at the bedside.
Alexandru G Floares
SAIA-Solutions of Artificial Intelligence Applications, Romania
Title: The impact of functional redundancy on molecular signatures
Time : 16:50-17:10

Biography:
Alexandru G Floares is a Medical Doctor (Neurologist) specialized in artificial intelligence applications in Oncology. Currently, he is the President of the Solutions of Artificial Intelligence Applications Organization and the OncoPredict Company. He completed his Neurology specialization and PhD in Biophysics at University of Medicine and Pharmacy, Iasi, Romania. His research project focus on “Developing non-invasive omics tests for cancer diagnostic, prognostic and treatment response prediction and automation of the mathematical modeling of complex biological networks with ordinary and partial differential equations using artificial intelligence”. He has publications and inventions in the fields above.
Abstract:
Precision medicine’s goals cannot be reached without discovering molecular signatures from omics big data, using machine learning. The prevailing conception is that for a given biomedical condition and class of biomolecules, there should be unique and highly accurate signature. Following the best practice for whet lab determinations and predictive models development, high accuracy (>95%) could often be achieved. However, the uniqueness request contradicts biological systems organization. They are amazingly robust, despite being highly complex. Evolution seems to favor functional redundancy which confers robustness. Until recently, we were searching for single relevant biomarkers. Then, we moved to single list of biomarkers. Now, we should move to multiple equivalent lists and even go beyond lists. Multiple different lists could be the results of different feature selection or modeling algorithms. If they are all highly accurate and relevant for the biomedical condition, they probably reflect functional redundancy. As most algorithms were developed with the goal of finding the smallest highly relevant subset of variables, they precluded the proper exploration of redundancy. That is why; we will have multiple equivalent small lists. Integrating many such lists could give a better representation of the biological reality. We can also go beyond the too simple concept of a single list with molecules increased or decreased in a particular condition. Using decision trees, one can obtain rules-sets containing biomarkers, thresholds and their conditions for each phenotypic class. This vision will be illustrated with our results from analyzing the biggest miRNA NGS data collection in cancer from TCGA. This work was supported by the research grants UEFISCDI PN-II-PT-PCCA-2013-4-1959 INTELCOR and UEFISCDI PN-II-PT-PCCA-2011-3.1-1221 IntelUro, financed by Romanian Ministry of Education and Scientific Research.
Magda Babina
Charité University Medicine Berlin, Germany
Title: The mast cell has much in store for us: A call to bioinformaticians to advance insights into a unique, intriguing but underexplored cell subset of the human body
Time : 17:10-17:30

Biography:
Magda Babina completed her PhD in Biochemistry in 1999 from Free University Berlin, Germany. She has studied Mast Cell Biology throughout her career, with emphasis on MCs in humans. She is a Senior Scientist and group Leader at Charite, Department of Dermatology and Allergy, and has published more than 50 papers. She has been a member of the FANTOM consortium (led by RIKEN in Japan) since 2010, one of the largest life science consortia in the world. This collaboration inspired her to obtain a more comprehensive and complete view of human MCs through systems biology approaches in collaboration with bioinformaticians.
Abstract:
Mast cells (MCs) are best known as effector cells of allergy but suspected to perform a range of other functions. Our knowledge of MCs in humans is seriously limited, as was one message from the collaborative endeavor FANTOM5 which used deep-CAGE sequencing on skin-derived MCs to generate a comprehensive view of their transcriptome. MCs were embedded in the body-spanning atlas, the datasets allowed to directly contrast their molecular signature against ≈200 primary cells. Our work demonstrates that: MCs are unique cellular elements; have no near neighbor; are intensely adaptable and; display transcriptional peculiarities. Our work also demonstrates: Uniqueness: MCs combine “private” with pan-hematopoietic genes supplemented by genes of disparate organs (e.g. neuronal/reproductive); Position: MCs have no close relative in the hematopoietic network being well separated from all other lineages, both by principal component analysis and by pairwise correlation analysis; plasticity: MCs show substantial adaptations regarding transcriptome, protein/mediator expression and functional programs in new microenvironments and; peculiarities: Cells with greatest TF diversity across atlas (893/MC versus 617/average) and many non-annotated transcripts exclusively active in MCs. Encouraged by these findings, novel functionalities of MCs have been uncovered (e.g. active BMP receptor and significance of the retinoid network) but burning questions remain such as “What is the nature of the TF network underlying lineage specification?”, “How do non-annotated transcripts contribute to MC identity?”, “How are genes from unrelated tissues activated in MCs”? Detailed bioinformatics analyses will help identify the most probable interconnections to facilitate further examination by wet-bench biologists.
Elena Papaleo
Danish Cancer Society Research Center, Denmark
Title: Structural communication in transcription factors: How interactions with DNA, mutations or post-translational modifications reshape the interfaces for cofactor recruitment
Time : 17:30-17:50

Biography:
Elena Papaleo completed her PhD in 2006 and Post-doctoral from 2007-2009 at the Department of Biotechnology and Bioscience at the University of Milano-Bicocca (Italy) in the group of Prof. Luca De Gioia and Prof. Piercarlo Fantucci. She was then appointed as Adjunct Professor in Computational Biology at the University of Milano-Bicocca from 2010-2012. Afterwards, she was Senior Post-Doctoral Researcher in the group of Prof. Lindorff-Larsen at the Department of Biology of the University of Copenhagen (Denmark) from 2011-2015. She has been Visiting Researcher at many international institutes including the group of Prof. Salvador Ventura at the Institute of Biotechnology and Biomedicine (IBB, Barcelona, Spain) and the group of Prof. Francesco Luigi Gervasio at the Spanish National Cancer Research Center (CNIO, Madrid, Spain). In August 2015, she joined as Group Leader of the Computational Biology (CBL) Laboratory at the Danish Cancer Society Research Center (Copenhagen, Denmark). She has authored more than 50 scientific papers as main or senior author and she is Academic Editor of PLoS One, Frontiers in Molecular Biosciences (Nature publishing group), PeerJ and Journal of Molecular Graphics and Modelling. The main research of CBL focuses on “Molecular modelling and simulations integrated with experimental data and network theory to the study structure-function relationship in key cancer proteins as well as on the analyses of high-throughput sequencing and omics data from profiling of cancer patients”.
Abstract:
Little is known about the molecular mechanisms related to the conformational changes induced at distal sites in many transcription factors which are often related to cancer disease, such as p53, MZF1 and the family of ARID domains. Thus, my group is focusing on the characterization of their structural dynamics to enrich the knowledge on this important group of regulatory proteins. In particular, we are employing a combined approach that integrates atomistic microsecond molecular dynamics simulations, enhanced sampling techniques, methods inspired by graph theory and cross-validation of the simulated ensembles with NMR data. To relate these properties to protein function, we studied both the free and DNA-bound forms of wild type, mutated and phosphorylated variants of p53, ARID proteins and MZF1. The interaction with DNA not only stabilizes the conformations of the DNA-binding loops, but also strengthens pre-existing paths in the free protein for long-range communication to potential interface for cofactor recruitment. Conformational states of these distal regions that are only a minor population of the free ensemble are promoted by DNA interactions, altering the preferences for certain classes of biological partners and thus influencing the signaling pathways mediated by these proteins. Moreover, mutations or post-translational modifications can also contribute to reshape the population of these interfaces even in domains that are not necessarily involved in DNA-binding.
Lee Wei Yang
National Tsing Hua University School of Life Science, Taiwan
Title: GNM 2.0- A database housing protein dynamics data of >11,000 PDB structures revealing dynamics prerequisites for protein functions and interactions
Time : 17:50-18:10

Biography:
Lee Wei Yang is currently an Associate Professor at Institute of Bioinformatics and Structural Biology, National Tsing Hua University, received his PhD degree in Molecular Genetics and Biochemistry from School of Medicine, University of Pittsburgh (2005) and his Post-doctoral training in University of Tokyo (2006-2009), La Jolla Bioengineering Institute and Department of Chemistry, Harvard University (2010-2011) before joining NTHU in 2011. He has published more than 30 papers in reputed journals and has an H-index of 16.
Abstract:
Proteins function through advantageously utilizing a repertoire of possible modes of intrinsic motions. Understanding such motions, of which the importance has been acknowledged in year 2013’s Nobel Prize in Chemistry, is essential to accurately predict two body interactions, channel gating mechanisms and enzyme catalysis. Here, we present the only dynamics database that houses dynamics data (vibrational normal modes) of protein structures in a size commensurate with Protein Data Bank (PDB). The interface that presents such data is state of the art of its kind. Given the wealth of the data, we are able to find dynamics traits for enzyme active sites. Such traits are later used to predict the locations of enzyme active sites for protein structures of a resolution as low as 20 Å. We further data-mined the database and develop the concept of intrinsic dynamics domains (IDDs), including a domain plane (D-plane) and a domain axis (D-axis). It is found that a protein interacts with another at the interface where D-plane cuts through and forming a near-vertical angle between two intersecting D-axes from the two proteins over a set of 68 protein–protein complexes. The findings are then used to define quantitative criteria to filter out docking decoys unlikely to be native whereby the chance to find near-native hits is doubled. Our results also show that in 95% of the DNA-protein complexes, the DNA is cut through by protein’s D-plane. The dynamics database, GNM 2.0, is made available at http://dyn.life.nthu.edu.tw/gnmdb and IDD website is provided at http://dyn.life.nthu.edu.tw/IDD/IDD.php.
Xiaoyu Yu
University of Pretoria, South Africa
Title: Mathematical modelling of evolution of tetranucleotide usage patterns of whole bacterial genomes to improve phylogenetic inferences
Time : 18:10-18:30

Biography:
Xiaoyu Yu has completed his MSc in Bioinformatics and is currently doing a PhD (2nd year) in Bioinformatics at the University of Pretoria. He has currently no publication with one in working progress and has attended and presented (posters) in many conferences in Europe such as ECCB 2014, ECCB 2015 and VAAM2016.
Abstract:
Nowadays, complete genome sequences of multiple bacteria became readily available for analysis. Current work which uses whole genome based alignment (WGS) approach for phylogenetic and phylogenomic research believe to resolve contradiction between gene based trees, but this approach multiplies the problem in terms of gene annotation, orthology prediction and inadequate alignment of sequences. Therefore one of the most prospective ways for genome comparison and phylogenomic inferences is then based on annotation-and-alignment free genome linguistic approaches, i.e. comparison of oligonucleotide usage patterns (OUP) of genome-scale DNA fragments. Until now, this approach still lacks a reliable evolutionary model to explain the mechanisms and dynamics of changes in OUP which hinders the application of this approach to systematically compare to other well-known methods such as marker genes and/or whole genome sequence based alignment. The aim of the current work is divided into three important topics: i) Comparative analysis of multiple complete genome sequences representing different phylogenetic branches at different taxonomic levels to identify the driving forces of OUP evolution; ii) Analysis of topological incongruences between phylogenetic trees based on orthologous gene alignments, whole genome alignments and alignment free OUP patterns; iii) Improving phylogenetic inference by reconciliation of sequence based and pattern based evolutionary models. The major output of this research is an innovative evolutionary model implemented in a form of a computer program for phylogenetic inferences based on combination of alignment based and alignment free approaches.
Matteo Lo Monte
Institute of Protein Biochemistry - CNR, Italy
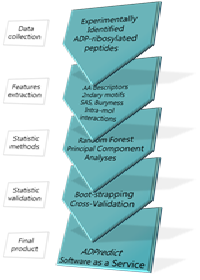
Title: ADPredict: ADP-ribosylation sites prediction based on physicochemical and structure descriptors
Time : 18:30-18:50

Biography:
Matteo Lo Monte completed his PhD in Medicinal Chemistry with a project entitled, “In silico screening of taste receptors: An integrate modeling approach” in 2015, at the “Universita’degliStudi di Milano”, under the supervision of Prof. Giulio Vistoli and in collaboration with Dompé Farmaceutici SpA, under Dr. Andrea Beccari. During this period, he mainly worked on the TRP receptors family, predicting their 3-D structure by homology modeling techniques and studying their interaction capacities as well as their activation mechanisms by Molecular Docking and Molecular Dynamic simulations; so generated models were conveniently utilized in virtual screening campaigns that successfully led to the identification of new hits. He moved to the Institute of Protein Biochemistry at the National Research Council in Naples in 2015 as Postdoctoral Fellow in the research group of Dr. Alberto Luini. He computationally supported several ongoing projects of Molecular Biology through molecular modeling and structural analyses studies of diverse enzymes like β4Galt5, SHIP1 and AGPAT4, as well as investigating the post-translational modifications, ADP-ribosylation in particular, developing predictive algorithms.
Abstract:
Statement of the Problem: ADP-ribosylation is a post-translational modification governing several crucial cellular processes, such as inflammation, cell survival or damaged DNA detection and repairing machinery activation. It is, thus, strictly related to neoplastic conditions. To date, ADP-ribosylation is poorly understood, as still incomplete is the knowledge of its effects on numerous molecular paths. Deeply understanding the circumstances in which it happens, as well as the numerous target proteins and the way their role in their respective biological pathways are affected by this event, would represent an important achievement in molecular biology, not solely for the progression in combating cancer.
Methodology & Theoretical Orientation: ADPredict is an in silico predictive algorithm of ADP-ribosylated Aspartate and Glutamate residues, based on both known physicochemical parameters (Z-Scales, ST-Scales, MSWHIM, ProtFP) and in-house derived secondary structure related and 3-D descriptors of hundreds of human ribosylated proteins. ADPredict was developed using principal component analyses (PCA) and the random forest algorithm. Its predictive capacity was then evaluated via intensive boot-strap approaches.
Findings: Here we present the first computational predictive tool able to individuate the Aspartate or Glutamate residues that are most likely to be ADP-ribosylated in a target of interest. Predictions can be achieved via single or multiple models (meta-model strategy), so allowing each time a tailored approach. It will soon be available as an online service at the website.
Conclusion & Significance: ADPredict arises as a new, concrete support to the study of the ADP-ribosylation event, flanking the analytic approaches developed so far and addressing the experimental investigation of this important biologic phenomenon. Ongoing extension of the predictive algorithm would aim to account also for the modification of additional amino acidic residues, so enlarging the applicability domain of the tool.
Jean-Didier Maréchal
Universitat Autònoma de Barcelona, Spain
Title: GAUDIMM: Widening the application of genetic algorithms for molecular modeling

Biography:
Jean-Didier Maréchal has completed a double PhD in chemistry from the Université Paris Sud (France) and Universitat Autònoma de Barcelona (Spain). He completed four years of Post-doctoral studies in England (Leicester and Manchester) and France (Paris Sud). He returned to Spain in 2006 and now leads his group on molecular modelling and design and presents a record of more than 65 papers in reputed journals.
Abstract:
Here, I present an optimitzation plateform for 3D molecular design based on a multi-objective genetic algorithm. GAUDIMM (Genetic Algorithm Under Design Inference and Molecular Modeling) is able to take several chemical descriptors (genes) at once and optimize them against a number of structural evaluators (objectives). Its aim is to provide with physically sound 3D models and its particularity is to allow the researcher to fill the geometric search with information generally partial provided by different sources, including data from other levels of theory, experiments or chemical intuition. The researcher only has to chose the adequate genes and objectives for his/her problem. GAUDIMM is provided with an API and the users can code their own extensions to adapt the optimization engine to their needs. With the already available built-in descriptors, one is able to select several molecules at once (think about competitive multiligand protein-ligand dockings), explore conformational flexibility (torsion angles and rotamers) and chemical variability (mutated residues, chemical group replacements). On the optimization side, a plethora of objectives have already been implemented from scoring functions to accurate force field energy calculations, simplified chemical interactions (hydrogen bonds, hydrophobic patches, steric clashes, covalent bond formation), geometric evaluation (distances, angles, surfaces areas, volumes, etc..) and metallic coordination. To date, several projects are challenged to tackle by standard computational methodologies have been successful thanks to GAUDIMM and include artificial enzyme re-design, geometries of metal-coordinated peptides and biosensors. While docking is not its main target, our platform even reports good benchmarks when tested against commonly used datasets.
Mahreen Arooj
Curtin University, Australia
Title: Structure-based systems biology for prediction of off-targets, biological pathways & target diseases

Biography:
Dr. Arooj has completed her PhD from Gyeongsang National University, South Korea in 2013. She has worked as Postdoctoral Research Fellow at Plant Molecular Biology and Biotechnology Research Centre (PMBBRC). Currently, she is working as Early Career Research Fellow in the School of Biomedical Sciences, Curtin University, Australia. Her research interests and expertise are to apply computational techniques such as structural bioinformatics, systems biology, and molecular dynamics in the fields of biomedical sciences.
Abstract:
Off-target binding connotes the binding of a small molecule of therapeutic significance to a protein target in addition to the primary target for which it was proposed. Progressively such off-targeting is emerging to be regular practice to reveal side effects. Here,we have developed a robust computational strategy using structure-based systems biology approach that is applicable to any enzyme system and that allows the prediction of drug effects on biological processes. Chymase is an enzyme of hydrolase class that catalyzes hydrolysis of peptide bonds. A link between heart failure and chymase is ascribed, and a chymase inhibitor is in clinical phase II for treatment of heart failure. However, the underlying mechanisms of the off-target effects of human chymase inhibitors are still unclear. In this study, putative off-targets for huamn chymase inhibitors were identified through various structural and functional similarity analyses and molecular docking studies. Finally, literature survey along with KEGG pathway maps was performed to incorporate these off-targets into biological pathways and to establish links between pathways and particular adverse effects. Off-targets of chymase inhibitors are linked to various biological pathways such as classical and lectin pathways of complement system, intrinsic and extrinsic pathways of coagulation cascade, and fibrinolytic system. Prospectively, our approach is helpful not only to better understand the mechanisms of chymase inhibitors but also for drug repurposing exercises to find novel uses for these inhibitors. This study also demonstrates the significance of computational strategies for efficacy prediction and the role that systems biology may play in multitarget therapeutics.
Alexander Zamyatnin
Russian Academy of Sciences, Russian Federation
Title: Fragments, fragmentome and fragmentomics in proteomics

Biography:
Alexander A. Zamyatnin is a physicist originally (M.V.Lomonosov Moscow State University, Physical Faculty), has PhD degree in physics and mathematics (biophysics), DSci degree in biology (human and animal physiology), Professor title (biophysics). Work: in different scientific organizations of several countries (Russia, USA, Hungaria, Chile, etc.). Studies: thermodynamics of muscle contraction; creation of the EROP-Moscow oligopeptide database; computer biophysics and biochemistry of structure and functions of the natural regulatory oligopeptides. He has more than 200 scientific publications, participates in the national (Russian and Chilean) scientific projects.
Abstract:
Natural fragmentation of biological molecules is well known. Fragmentary structural organization is characteristic of both the simplest and most complex biological molecules. Low molecular weight fragments of biological substances can be easily seen on metabolic maps. Therefore the term “fragmentomics” is grounded and defined, the bases and determination are given for the notion of the “fragmentome” as a set of all fragments of a single substance, as well as for global fragmentome of all chemical components of living organisms. A steady increase in the number of publications dealing with protein fragment has been seen in recent years. For some proteins there are already hundreds of fragments that have been studied in detail, and it seems that concepts concerning functional importance of the totality of possible fragments of a single protein will be formed. For peptide structures, fragmentomics can be considered as a notion that combines proteomics and peptidomics. EROP-Moscow (Endogenous Regulatory OligoPeptides) database demonstrates structural and functional variety of possible protein fragments. Formation of an exogenous–endogenous pool of oligopeptides in an organism and correlation of these data with concepts of structure–functional continuum of regulatory molecules is shown on an example of milk and meet protein fragments.

Biography:
Abstract:
Several studies showed the importance of rhizobacteria (P. fluorescens) associated to natural fertilizer improving cereal production in poor and arid soils. However, agriculture crops prediction by statistical models remains an adequate system to assess the performance of bio-fertilizer used in farming practices. Indeed, the large amount of data to process in this context allowed the integration of computers in the statistical analysis schemes. Computational statistic can be defined as the explicit impact of computers on statistical methodology. Here,we developed a bioinformatics pipeline in R bio-statistic environment assessing the relationship between previous analyzed rhizobacteria (P. fluorescens) treatments (T0: treatment without any rhizobacteria and any foliar bio-fertilizer, T1: treatment with only rhizobacteria, T2: treatment with both rhizobacteria and foliar bio-fertilizer and T3: treatment with only foliar bio-fertilizer)and their potential influences on growth and yield parameters of both maize and soybean cereal varieties in arid soil in the north of Côte d'Ivoire. Then, the present survey basing on the computational statistic approach, highlighted a strong difference between the four considered rhizobacteria treatments impacting the two analyzed cereal crops and development process (p-values < 0.05). Moreover, the same analysis suggested a positive and selective effect of rhizobacteria (P. fluorescens) combined with foliar bio-fertilizer on both quantitative and qualitative production of analyzed cereal crop varieties. Indeed, we were able to demonstrate the differences between maize and soybean crops replying torhizobacteria (P. fluorescens) bio-fertilizer treatments. Further, the present developed pipeline showed that the two analyzed varieties of soybean (green and yellow soybean) were differentially influenced by the different rhizobacteria treatments as opposed to maize plant varieties (p-value < 0.05). Finally our findings evidenced that disregarding analyzed parameters and cereal varieties, treatment T2 having the recommended dose of rhizobacteria P. fluorescens+ foliar fertilizer compost recorded the best performance improving both maize (Zea mays. L) and soybean (Glycine max) cereals cultivation in arid region. In conclusion this study demonstrated the key role of rhizobacteria (P. fluorescens) combined with foliar bio-fertilizer improving cereal production in soil with low fertility aptitude, adjusting the concordance between both growth and yield parameters.
- Track 4: Bioinformatics Algorithms & Databases
Track 5: Immunology and Drug Discovery
Track 6: Perspectives of Clinical Informatics

Chair
Nabil Mili
University of Geneva, Switzerland

Co-Chair
Tamas Hegedus
Semmelweis University, Hungary
Session Introduction
Maria Vittoria Cubellis
University of Naples, Italy
Title: Workshop on Disease specific databases for personalized medicine: Fabry disease as a case of study
Time : 14:20-15:05

Biography:
Maria Vittoria Cubellis graduated in Chemistry from the University of Naples and obtained the PhD in Biochemistry. She is professor of Biochemistry and Bioinformatics at the University "FedericoII" of Naples. Her recent research interests are focused on pharmacological chaperones for the treatment of rare diseases and in particular on the prediction of mutations associated to Fabry disease which are responsive to drugs. She is also trying to extend the approach with pharmacological chaperones to other pathologies, such as disorder of glycosylation type 1a, a disease for which there is no cure at present.
Abstract:
Next generation sequencing of all exons will become common also for symptomless people in the near future. Since we all have approximately 20000 variants, differentiation among non-pathological, mild or deleterious mutations will be necessary. We will show how disease specific databases and predictive tools can be precious for personalized diagnosis and therapy. The analysis of sequence conservation among orthologous proteins, even in the absence of structural information on the human protein, can be sufficient to identify severely pathological mutations, but training sets represent the weak point in the development of prediction tools. At present more than 70000 missense mutations have been reported, with 7 variants per protein on average, but at least 70 cases more than 100 variants are known. A quantitative phenotype can be associated to mutants measuring the residual activity or the stability of proteins expressed by transient transfection. In this case it can be possible to predict the severity of the mutation quantitatively. This implies that in many cases it is possible to develop disease specific predictive tools. Lysosomal alpha galactosidase, which is associated to Fabry disease, represents a good case to test the effectiveness of specific tools that allow predictions scored according to severity. More than 380 missense mutations are known and for 305 the residual activity in cells has been assessed. For the deficiency of lysosomal alpha galactosidase, disease specific predictive tools can also be exploited to estimate responsiveness to specific drugs such as pharmacological chaperones.
Nabil Mili
University of Geneva, Switzerland
Title: Translating genomic biomarker findings into clinical medecine: Dimension of the problem, network structure of competing models and estimate of the misclassification prediction error
Time : 15:05-15:25

Biography:
Nabil Mili completed his Doctorate in Medicine and MSc in Statistics from the University of Geneva. He is board certified in Anesthesiology (Switzerland). His domains of interest are “Model selection in high dimensional data and the connection between statistical formalization and medical reasoning”. He is currently working in the Research Center for Statistics-University of Geneva.
Abstract:
Technical breakthroughs have enabled unprecedented progress in the field of omics. Arguably, this should result in great potential in the field of biomarker discovery and indeed, publications in the field of biomarker discovery have increased dramatically over the past two decades. However, the increase in the number of clinically useful biomarkers have been meager. The major statistical challenges in the translation from biomarker discovery to clinical utility, as long as the concern is to classify any patient into a nosographic category are: The dimension of the problem (how many biomarkers do we need to properly classify a patient?); the network structure of the selected biomarkers (a biomarker does not act in an isolated way but is inserted into a network. What is the architecture of that network?); the paradigmatic structure of competing statistical models (many rival models may have the same misclassification predictive error. They should be considered as equivalent or belonging to the same paradigmatic class) and; an estimate of the misclassification prediction error. We applied a newly proposed gene selection method based on statistical and machine-learning principles which delivered a set of models that best predicted the disease class. These models were inserted in a network where the biomarkers were placed in specific positions according to their relevance in discriminating between the diseases. The principles of the method that meets the above mentioned challenges will be presented and applied to clinical cases (inflammatory bowel diseases, breast cancer and gliomas).
Patrick Kück
The Natural History Museum London, UK
Title: PhyQuart-A new algorithm to avoid systematic bias & phylogenetic incongruence: Are directed quartets the key for more reliable supertrees?
Time : 15:25-15:45

Biography:
Patrick Kück has completed his PhD at the Zoological Research Museum A. Koenig and the Rheinische Friedrich-Wilhelm University in Bonn, Germany, with research focus on the development of new algorithms for data evaluation and homology assessment in phylogenetic reconstructions. He is currently an IEF Research Fellow at the Natural History Museum London, UK. He has published more than 20 papers in reputed journals.
Abstract:
Recent phylogenetic studies of old taxonomic relationships point out how sensitive probabilistic tree reconstruction methods are to the selection of model assumptions and data compositions. Systematic errors are characterized by getting increasingly apparent, the more data are analyzed. An alternative to phylogenetic reconstruction of complete data sets is the divide and conquer principle which divides overall reconstruction problems into smaller subsets. The phylogenetic information gained from such subset analyses is subsequently used to generate a phylogenetic supertree comprising all taxa. Quartet based methods are very attractive for supertree reconstructions. A quartet topology comprises the phylogenetic information inferred from a set of four taxa sequences and is the smallest phylogenetic informative unrooted tree. This talk presents PhyQuart, a new algorithm based on a site pattern classification for quartets of aligned sequences using observed and expected split-supporting site-patterns, considering two different topological directive transformations for the inner branch of each quartet relationship. Simulation analyses show that the combination of site pattern- and Maximum Likelihood analysis leads to quartet inferences that are nearly as good or in many cases even better than in conventional ML-analyses, especially with strong rate heterogeneity. First tests in combination with a new developed supertree technique suggest that PhyQuart might be a good alternative to reduce systematic bias in quartet-based divide and conquer approaches.
Alvaro Olivera-Nappa
University of Chile, Chile
Title: VHL-Hunter: A predictor of the impact of missense mutations in the function of the Von Hippel-Lindau protein (pVHL), risk of clear cell renal carcinoma and severity of pVHL-associated diseases
Time : 15:45-16:05

Biography:
Alvaro Olivera-Nappa has received a PhD in Chemical Engineering, specializing in Biotechnology and Protein Engineering. He was a Post-doctoral Fellow in Santiago, Chile, and Delft, The Netherlands, and has occupied a Visiting Scholar position at the Department of Biochemistry of the University of Cambridge, UK, under Professor Sir Tom Blundell. Since 2013, he is an Assistant Professor at the University of Chile. He has published articles in the fields of protein science, protein engineering, mathematical modeling of biological systems and the prediction of the effect of mutations in cancer and disease. He has developed mathematical, statistical and computational tools to understand and design protein function from a molecular and relational point of view, to represent and analyze complex chemical and biological processes and to design and implement biotechnological solutions and applications. He is interested in biotechnology applications for economic development and constantly promotes innovation and science education as drivers of social development.
Abstract:
Von Hippel-Lindau (VHL) disease is an autosomal dominant syndrome associated with multiple tumors including hemangioblastoma, clear cell renal carcinoma (ccRCC) and pheochromocytoma (PCC), which results from mutations in the VHL gene. VHL disease is classified into type 1 or type 2 depending on the presence or absence of PCC. A major limitation is that accurate classification can only be made in large kindreds. Furthermore, its use in assisting clinical management is limited since a family may move from one subtype to another. More than 600 VHL disease mutations are missense, which are broadly distributed throughout the gene. Inheritance of VHL mutations in an autosomal recessive fashion can lead to congenital polycythaemias. Germline VHL mutations account for up to 50% of patients with apparently isolated familial PCC and 11% of patients with an apparently sporadic PCC. Additionally, somatic biallelic inactivation of VHL also occurs in the majority of sporadic ccRCCs, with ~250 different missense mutations described. Numerous studies have investigated with conflicting results whether partial/total loss of VHL function or the type of VHL mutation may influence prognosis in ccRCCs. Our group previously developed symphony, a predictor of ccRCC risk associated to VHL mutations. In this work we present VHL-Hunter, a new computational approach to predict all VHL-associated disease risks, which combines structural and H-bond network induced pVHL amino acid partitioning, MOSST and SDM predictions. VHL-Hunter correctly predicts the disease outcome of known pVHL mutations with high accuracy (0.96) and informedness (0.9), and also the outcome of previously undescribed mutations. VHL-Hunter also gives clues regarding the mechanism underlying disease generation and is able to correlate the predicted degree of protein malfunction with the severity of clinical phenotypes, in agreement with experimental data. This method can also be successfully applied to obtain highly accurate predictors of mutation phenotypic outcomes for other proteins
Runsheng Chen
Institute of Biophysics, Chinese Academy of Sciences, China
Title: Big Data in noncoding RNA and precision medicine
Time : 16:05-16:25

Biography:
Runsheng Chen, Principal Investigator at Institute of Biophysics, CAS, is an Academician of the Chinese Academy of Sciences (CAS) and an Academician of the International Eurasian Academy of Sciences. He is a member of Human Genome Organization (HUGO), a member of the biomacromolecule group of the Committee on Data for Science and Technology (CODATA) and a member of the bioinformatics professional committee of the International Union of Pure and Applied Physics (IUPAP). He is now the General Secretary and Vice President of Chinese Society of Biophysics and has published more than 130 papers in SCI.
Abstract:
The living organisms on the earth from prokaryotes to eukaryotes have been proliferating for billions of years. To date, they form in more complicated structure and function in more perfect ways. However, what really determined the complex phenotype, structure and function of living organisms? Where do they store those huge amounts of information? And how do they operate? All of these have been keen questions for people to explore. What has been astonishing and puzzle is the fact that life is not just a simple group of molecules instead, it is highly organized. There is connections between nucleus and cytoplasm, a clearly work division between different organizations and synergy cooperation within organs. Therefore, a normal living organism is extremely multi-level and dynamic. The complexity of the organism is not only reflected in the complexity of the structure of DNA information but also on the implementation of the information and operation rule. This report mainly introduces the rise of noncoding area, great innovation opportunity it offers and the role of big data in this field. Meanwhile, this report also introduces what the scientists has been explored for the associations between genotype and phenotype. As a result, series of new concepts such as translational medicine, personalized medicine and precision medicine etc. have been put forward by medical scientists. All of these imply that the big changes for medical system from diagnosis and treatment to health care are upcoming. It also suggests the birth of a new generation of huge health care industry.
Tamas Hegedus
Semmelweis University, Hungary
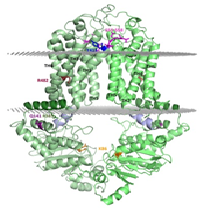
Title: In silico modeling of ABCG2/BCRP structure and dynamics sheds light on xenobiotic recognition and transport function
Time : 16:45-17:05

Biography:
Tamas Hegedus has been a graduate student in the laboratory of Balazs Sarkadi (NIHI, Budapest, Hungary) studying ABC proteins. He has completed his Postdoctoral studies on CFTR/ABCC7 from the laboratory of Jack Riordan (Mayo Clinic Scottsdale, AZ and UNC at Chapel Hill, NC, USA). He leads a group aiming to understand multidrug recognition and folding problem of CFTR mutants (www.hegelab.org). He is a board member of the Hungarian Society for Bioinformatics and a member of the Disease Database Advisory Council, Human Variome Project.
Abstract:
ABCG2 is an ATP binding cassette transporter protein, containing a nucleotide binding domain and a transmembrane region. It functions as a homo-dimer multidrug resistance (MDR) transporter in the plasma membrane, extruding a wide range of molecules from the cell. ABCG2 affects the pharmacokinetics of various drugs and protects the stem cells and cancer (stem) cells from xenobiotic and chemotherapeutic agents. This transporter is present in key pharmacological barriers and drug metabolizing organs. However, ABCG2 is also involved in the transport of endobiotics, e.g. porphyrins and uric acid. Its Q141K variant exhibits decreased functional expression leading to increased drug accumulation and decreased urate secretion. To overcome MDR and correct ABCG2 related pathological phenomena, learning its structural and dynamic properties is a major objective in the field. Still, there has been no reliable molecular model available for this protein, as the published structures of other ABC transporters could not be properly fitted to the ABCG2 topology and experimental data. The recently published high resolution structure of a close homologue, the ABCG5/ABCG8 heterodimer, revealed a new ABC transporter fold, unique for ABCG proteins. We generated a structural model of the ABCG2 homo-dimer based on this fold and confirmed its validity using experimental data. We also performed molecular dynamics simulations and in silico docking to understand the effect of mutations including R482G and Q141K and the mechanism of substrate recognition. The ABCG2 model, we present may have significant impact on understanding drug distribution and toxicity as well as drug development against cancer chemotherapy resistance or gout.
Surya Raj Niraula
B P Koirala Institute of Health Sciences, Nepal
Title: Probability sampling in matched case-control study in drug abuse
Time : 17:05-17:25

Biography:
Surya Raj Niraula has completed his PhD from Tribhuvan University and Post-doctoral from University of Washington, USA. He is the Professor of Biostatistics at B P Koirala Institute of Health Sciences, Nepal. He has published more than five dozen of papers in reputed journals and has been serving as a Statistical Reviewer in many national and international journals. He was awarded with ‘Young Scientist Award’ in 2009, Nepal Bidhyabhusan KA in 2010 and Honored by the President - Constitutional Assembly, 2011. He has presented many conference papers in USA, UK, Korea, Singapore, Thailand, India and Nepal.
Abstract:
Although random sampling is generally considered to be the gold standard for population-based research, the majority of drug abuse research is based on non-random sampling despite the well-known limitations of this kind of sampling. We compared the statistical properties of two surveys of drug abuse in the same community: one using snowball sampling of drug users who then identified “friend controls” and the other using a random sample of non-drug users (controls) who then identified “friend cases”. Models to predict drug abuse based on risk factors were developed for each data set using conditional logistic regression. We compared the precision of each model using bootstrapping method and the predictive properties of each model using receiver operating characteristics (ROC) curves. Analysis of 100 random bootstrap samples drawn from the snowball-sample data set showed a wide variation in the standard errors of the beta coefficients of the predictive model, none of which achieved statistical significance. On the other hand, bootstrap analysis of the random-sample data set showed less variation and did not change the significance of the predictors at the 5% level when compared to the non-bootstrap analysis. Comparison of the area under the ROC curves using the model derived from the random-sample data set was similar when fitted to either data set (0.93 for random-sample data vs. 0.91 for snowball-sample data, p=0.35); however, when the model derived from the snowball-sample data set was fitted to each of the data sets, the areas under the curve were significantly different (0.98 vs. 0.83, p<.001). The proposed method of random sampling of controls appears to be superior from a statistical perspective to snowball sampling and may represent a viable alternative to snowball sampling.
Veronna Marie
University of KwaZulu-Natal, South Africa
Title: Structural implications of protease mutations in antiretroviral treatment exposed patients infected with HIV-1 subtype C
Time : 17:25-17:45

Biography:
Veronna Marie is pursuing her PhD at the HIV Pathogenesis Programme, Nelson R Mandela Medical School in Durban, South Africa. Her current research focuses on “Structural bioinformatics in HIV-1 drug resistance”. The aim of her current work is to “Find mutational pathways in HIV-1 subtype C that can translate into improved medical outcomes for patients failing therapy in resource limited settings”. Her previous research fields included Environmental Biotechnology and Water Research.
Abstract:
The development of drug resistance mutations (DRMs) within protease often results in therapy failure. Currently, limited studies have focused on the structural implications of DRMs in subtype C. The study aim was to assess structural changes within protease that may provide insight into mutational pathways in subtype C. All protease subtype C sequences were retrieved from public databases. After quality assurance, 1912 sequences remained. Resistance associated mutations were identified via the HIVdb algorithm. Phenotypic variation (PV) was calculated using the ConSurf server. Homology models were predicted using Modeller. High PV indicative of drug pressure corresponded to the sheets, 30s loop, core and active site regions of protease. Interestingly, 296 unique combinations of DRMs occurred; the most common was M46I+I54V+V82A (n=27). Combinations of three or more occurred frequently suggesting that these combinations were not random but might have structural implications. M46I+154V+V82A showed greater distances between the mutated residues in comparison to the wild-type (WT). M46I+I54V+V82A+L76V was also common (n=14). V32I, a darunavir mutation directly interacted with L76V and V82A, moving inhibitors away from I84 and towards I50. I54L, also a darunavir mutation, increased the distance of the hydrogen bonds between the mutant and I47 to twice (4.286 Å) that of the WT (I54+I47), modifying flap flexibility and active site dynamics. The interaction of L76V with darunavir mutations V32I, I54L and I84V may have implications when switching to darunavir after lopinavir failure. Understanding DRM pathways in protease can help to prevent therapy failure and promote drug design for next generation protease inhibitors.
Prakruthi Appaiah
CSIR-Central Food Technological and Research Institute, India
Title: Designer protein enriched with large neutral amino acids: A new approach for treating phenylketonuria
Time : 17:45-18:05

Biography:
Prakruthi Appaiah has completed her MSc in Microbiology (2010) from University of Mysore and currently pursuing her PhD (Life Science) in CSIR-Central Food Technological Research Institute, Mysore, India. After her Post-graduation, she worked as Project Assistant (2011) in Lipid Science and Traditional Food Department, CSIR-CFTRI, Mysore, during which she has published three papers in reputed journals and won Best Oral Presentation Award in the National Conference on Functional Foods in Health & Well-being, Bangalore, India. She has presented a poster in 22nd Indian Convention of Food Scientists and Technologists (ICFOST-2012), organized by AFSTI at CSIR-CFTRI, Mysore, India.
Abstract:
Phenylketonuria (PKU) is a genetically inherited disease caused by the defective phenylalanine hydroxylase (PAH) enzyme. In case of phenylketonuria, the body fails to convert phenylalanine (Phe) to tyrosine (Tyr), resulting in the elevated blood Phe level and consequent neurological damage. Of all therapies, large neutral amino acid (LNAA) supplementation has emerged as a promising approach for the dietary treatment of PKU. The LNAAs compete with Phe for the same L-type LNAA transporter (LAT1, SLC7A5) across the blood-brain barrier, decreasing brain Phe level. Thus, the aim of this study was to design an easily digestible protein enriched with LNAA (except Phe) in accordance with WHO/FAO/UNU specification by homology modeling using αs1 casein as template. The challenge was to maximize the LNAAs content (except Phe) in the protein model by finding a suitable scaffold (like α-helix) for homology modeling. Out of 63 different protein models designed, protein model-54 was selected for its compact 3D structure with only α-helices, high sequence similarity with template (60.4%) and good in silico digestibility. Different software like, SWISS-MODEL, EXPASY tool, PROFUNC, I-TASSER, RaptorX and SAVeS Server were used for the structure prediction and validation of the designed protein. The structures obtained from tertiary predicting software were visualized by discovery, UCSF chimera and Pymol tools. Based on these evaluations, the protein model-54 was found to be the best and reliable model. The presentation will review the strategies used for homology modeling, secondary structure and tertiary structure prediction and validation of the designed protein and discuss its nutritional significance for PKU treatment.
Omar Hussein Salman
Al Iraqia University, Iraq
Title: Remote patient triage and prioritization in mobile telemedicine services
Time : 18:05-18:25

Biography:
Omar Hussein Salman completed his graduation from Gifted Secondary in Baghdad with Diploma in 2001. Then, he joined Al Nahrain University, Baghdad and completed his BSc in Computer Engineering in 2004. He completed his MSc in Computer Engineering in 2007. In 2008, he joined the Iraqi University in Baghdad. He completed his PhD in Computer Systems Engineering from University Putra Malaysia in 2016 and his research was focused on “Multi-sources data fusion processing in telemedicine”. During his PhD. he got patent and published a research article. His research interest includes “Data processing, algorithms, remote applications, bioinformatics, telemedicine and healthcare services”.
Abstract:
The more the worldwide population gets older, the bigger is the need for technologies to monitor and assist patients in healthcare applications. Consequently, in order to accommodate the increasing number of users, the remote patient monitor one of the issues that telemedicine, mobile technology and Wireless Body Area Networks (WBAN) have to tackle on, and it constitutes the main focus of this research. To provide healthcare services for a huge number of users, the healthcare providers triage the patients. Triaging involves an initial sorting of patients in order to prioritize the most emergency patients and to ensure by providing them the appropriate and rapid healthcare services. This study proposes a framework to improve the remote triaging and remote prioritization processes for the patients who are in places that are far from the ED and with no triage nurse. The proposed framework named Multi Sources Healthcare Architecture (MSHA) considers multi-heterogeneous sources: Sensors (ECG, SpO2 and Blood Pressure) and text-based inputs from mobile and pervasive devices of WBAN. Simulation results based on datasets for different symptoms of heart diseases demonstrate the superiority of MSHA algorithms as compared to benchmark algorithms in terms of triaging and prioritizing patients remotely in healthcare applications.
Afsheen Mushtaque Shah
University of Sindh, Pakistan
Title: Hematological Abnormalities in patients with Active Pulmonary Tuberculosis

Biography:
She has completed her Ph.D at the age of 34 years from Sindh University, Jamshoro, Sindh, Pakistan. Working as teacher since 2002 and presently working as Associate Professor at Institute of Biochemistry, University of Sindh, Jamshoro, Sindh, Pakistan. She has published 14 research papers in research journals.
Abstract:
Mycobacterium Pulmonary Tuberculosis (PTB) is an enduring infectious disease of the world. Pakistan is a developed nation where the prevalence of pulmonary TB increases and graded as 5th mid of 22 TB elevated countries and is the second leading cause of death in the world. As a risk factor of PTB, tobacco smoking has increased substantially over the past three decades, especially in developing countries. Multidrug-resistant TB (MDR-TB) has become a significant public health problem. Abnormalities in hemoglobin, hematocrit levels and Erythrocyte Sedimentation Rate (ESR) are usually disturbed in infectious disease. The aim of this study was to determine the hematological changes in active pulmonary TB patients and compare with normal healthy control subjects. Total of 338 patients of both genders age ranged 20 to 60 years, with positive sign and symptoms of tuberculosis were selected from Institute of Chest Disease, kotri, Sindh, Pakistan, Liaquat University of Medical & Health Sciences, Jamshoro, Sindh, Pakistan, Liaquat University Hospital, Hyderabad, Sindh, Pakistan, & Rajputana Hospital, Hyderabad, Sindh, Pakistan. Blood was collected from each subject and sent to the research laboratory. Informed consent was taken prior to collection of sample. Our study was based on two groups. One was the Refamipicin sensitive PTB patients (RSPTB) and the other was Refampicin Resistant PTB patients (RRPTB) The mean ages of both gender of RSPTB and RRPTB patients were 36.7±14.1 and 34.0±13.9 years. The male and female patients of RSPTB were 94 and 70 while in RRPTB the male and females were 100 and 74. In RRPTB, the hematological parameters including RBC, PCV, MCH, MCHC, WBC N, L, M,E, PLT & ESR (4.2 ± 0.5), (32.6±5.1),(26.8±3.8), (31.0±1.4), (1.3±3.5),(82.4±9.2), (16.2±6.2), (11.6±5.5), (7.1±1.0), (3.4±1.4) & (63.3±31.1) were significantly higher and MCV (73.6±12.4) were significantly lower. While in RSPTB (3.7±0.7), (30.6±4.7), (23.1±4.0), (29.8±3.2), (1.2±4.2), (81.6±6.1), (16.98±7.5), (8.99±6.3), (5.1±1.8), (3.4±1.3), & (42.5±43.2) were significantly lower and MCV were (76.4±11.1), were significantly higher. As compare to control group all the biochemical parameters were statistically significant. It is conclude that hematological abnormities have a significant role in cure of patients suffering from active pulmonary tuberculosis.
Yanhong Gao
Chinese PLA General Hospital, China
Title: Sensitivity of Palb2-null tumor cells to an oxidative stress inducing agent

Biography:
Yanhong Gao has completed her PhD from Chinese Academy of Medical Sciences in 2005. She work as an associate chief physician and associate professor at Department of Clinical Biochemistry of Chinese PLA General Hospital since 2005. She is interested in finding tumor new biomarkers for diagnoses and prognostic from blood by advanced technology methods. She has published more than 25 papers in journals.
Abstract:
PALB2 gene mutations, as the newly discovered breast cancer associated gene, has brought new direction for the prevention and treatment of breast cancer. To better understand the function for PALB2 and whether it can be use for drug, we generated p53-single-null (as control) and Palb2; p53-double-null cell lines from the mouse mammary tumors obtained and we found the Palb2-null tumor cells were hypersensitive to DNA damaging agents in previous study. To explore new ways to selectively kill Palb2-null tumor cells, we tested the potential of targeting oxidative stress in the cells. For this purpose, we chose phenethyl isothiocyanate (PEITC) and L-sulforaphane. We tested the sensitivity of 5 different Palb2-null tumor cell lines and 3 different control lines to the drugs. Cells were seeded in 96 well plates and treated with different concentrations of the two drugs for 72 hr. Then, cell viability was measured by CellTiterGlo® assay. Comparing with L-sulforaphane, we found Palb2-null tumor cells were hypersensitive to PEITC. PEITC,a natural compound that is rich in vegetables such as watercress and broccoli, etc. PEITC has long been known to possess anti-cancer activity, has been extensively studied. According the result, it is raising an tempting prospect of preventing or treating PALB2-associated cancers with the inexpensive drug.
Biography:
João Lídio da Silva Gonçalves Vianez Junior is graduated in Biological Sciences at the UFRJ, MSc in Plant Biotechnology at the UFRJ and PhD in Biological Sciences (Biophysics) at the same institution. Currently is general coordinator of the Center for Technological Innovation of Evandro Chagas Institute (IEC) and coordinator of the Bioinformatics Core. It has experience in assembly, annotation of genomes and transcriptomics data from NGS platforms. He has knowledge in molecular phylogeny of microorganisms. It also has experience in modeling, molecular dynamics and anchoring.
Abstract:
Brazil is facing an unprecedented growth in the number of microcephaly cases in babies. This phenomenon coincided with the recent Zika virus (ZIKV) outbreak in this country. Although the Brazilian Ministry of Health was quick to recognize that ZIKV was probably the cause of microcephaly in newborns, the underlying mechanisms leading to the development of this pathology have not been established. To tackle this problem at the molecular level, we employed whole transcriptome sequencing of human neurospheres derived from neural stem cells exposed to ZIKV isolated in Brazil (Asian genotype). Differential gene expression analysis of control (MOCK) and ZIKV infected neurospheres generated a list of 26 down-regulated and 64 up-regulated genes. Among the up-regulated detected genes, the Cyclin-dependent kinase inhibitor 1A (CDKN1A) and the Glial fibrillary acidic protein gene (GFAP) were found. CDKN1A prevents the activation of the Cyclin E/CDK2 complex, acting as a regulator of cell cycle progression during G1 and GFAP is a known marker of astrocytes. We also observed a decrease in the expression of the neurogenic differentiation 1 gene (NEUROD1), which is directly involved in the neurogenic program.Those findings suggest that ZIKV infection induces cell cycle arrest and inhibits the neuronal differentiation, resulting not only in the reduction of the size, but in a deeper disruption of the normal development of the human brain.

Biography:
Dr. P. Ananda Gopu has received his Ph.D. degree in Biotechnology (subject bioinformatics) from Periyar Maniammai University, India in 2012. He has worked as Research Associate at Bioinformatics Research Centre (BIRC) of Nanyang Technological University, Singapore in 2009.08- 2011.03. Dr. Ananda Gopu has joined as Scientist at Centre for Research and Development PRIST University in 2014 and his goal is several applications in health care involved in understanding complex relationship between sequence, structure, functions and interactions of protein-protein & protein-DNA using computational analysis of biological data. He has 7 papers in peer-reviewed international and national journals to his credit
Abstract:
In the present study, the protein sequences have been preconceived in series of atoms instead of amino acids (AA). Specifically, the stretches of protein sequences have been considered preferably in terms of carbon atoms instead of using an AA-based pattern. Accordingly, we have observed that patterns consisting of the same number of carbons can result in different AA lengths with different numbers of total atoms. The variation of these patterns is characterized by a change in the distribution of large hydrophobic residues (LHRs) (such as phenylalanine, isoleucine, leucine, methionine and valine) with the introduction of higher numbers of small hydrophobic residues (SHRs), such as glycine, alanine, proline and cysteine. Consequently, proteins that have the same number of carbons can have different numbers of AA within their various patterns and thus an increase in their peptide length. Hence, proteins of the same carbon contents may show different patterns with different peptide lengths that may reflect their specific biological functions. Concluding, an atomic level comparison of protein sequences can provide better results than a similar comparison at the residues level which may have potential implications for the prediction of misfolded proteins.
- Workshop on Disease specific databases for personalized medicine: Fabry disease as a case of study